- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2014
- X-ray Imaging
- Why lung ventilation is unevenly distributed in asthma: new insight from synchrotron K-edge subtraction imaging
Why lung ventilation is unevenly distributed in asthma: new insight from synchrotron K-edge subtraction imaging
Asthma, the most common chronic disease in children, is characterised by chronic inflammation of the airways. Exposure to an allergen in sensitised asthmatics triggers the expression and release of mediators including cytokines, chemokines, adhesion molecules, inflammatory enzymes and receptors by inflammatory cells, which leads to bronchial smooth muscle constriction and an increased resistance of the bronchi to air flow. Allergen exposure also causes transient increases in eosinophils, T-cells, mast cells and neutrophils in the lung and inflammatory cell infiltration of airway walls. In the Brown Norway rat, a strain genetically predisposed to allergic hypersensitivity, allergen provocation produces patchy inflammatory cellular infiltrates in the walls of both airways and pulmonary blood vessels. Eosinophils are thought to play a central role in allergic inflammation in asthma, however, the relationship between eosinophilic infiltration and regional lung function is poorly understood.
Bronchoconstriction in asthmatic subjects leads to a significant heterogeneity in the distribution of inhaled air within the lung, and the appearance of ventilation defects [1]. This phenomenon is important not only because it contributes to abnormal respiratory mechanics and gas exchange, but also because it can significantly impact the distribution of inhaled drugs commonly used in the treatment of asthma. We have previously shown that both the route (inhaled vs. blood-born) and nature (allergic vs. non-specific) of airway challenge play a significant role in determining the heterogeneity of airway constriction [2]. However, the exact mechanisms causing such striking regional differences in lung ventilation in allergic asthma remain elusive.
We studied the relationship between the spatial distribution of the cellular effectors involved in asthma and the emergence of defects in regional ventilation following allergen exposure. For this purpose, we used in vivo high-resolution K-edge subtraction (KES) synchrotron imaging at the ID17 beamline, during xenon inhalation in ovalbumin (OVA)-sensitised Brown-Norway rats at baseline and repeatedly following OVA challenge.
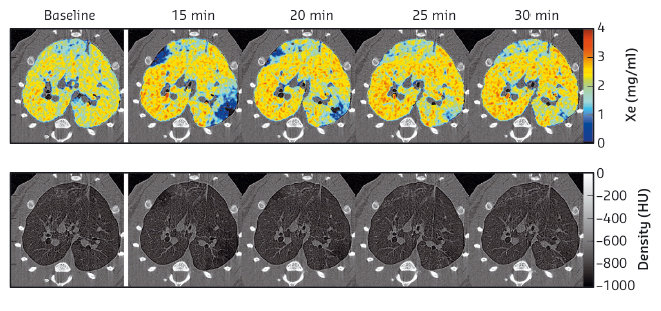
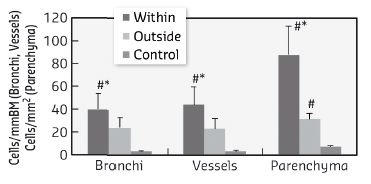
The KES technique allows simultaneous imaging of the airway structure and quantitative measurements of the regional tissue density and the concentration of inhaled Xe gas [2]. This technique allowed us to measure both airway narrowing and the distribution of inhaled Xe gas within the pulmonary alveoli. Histological slices of the lung anatomically matched to the computed-tomography images were stained and digitised. Total cells, CD68+ and eosinophils were counted in the walls of bronchi and vessels randomly selected within and outside of ventilation defects, delineated based on the Xe-KES images. Following OVA challenge, we observed transient ventilation defects (Figure 63), and increases in airway resistance and tissue elastance. Eosinophil (Figure 64), CD68+ and total cell counts were significantly higher in the bronchial and vascular walls within vs. outside of the ventilation defects. Furthermore, airway constriction during OVA-induced bronchoconstriction was correlated with eosinophil and total cell densities in the airway walls within the poorly-ventilated zones.
 |
|
Fig. 63: High resolution K-edge subtraction images for one representative animal. Upper row: quantitative Xe-images. Following OVA, transient clustered areas of poor ventilation emerge (blue). Lower row: tissue-density KES images. |
 |
|
Fig. 64: Eosinophil cellular densities in airway wall, vessel wall and parenchyma, within and outside the ventilation defects, defined based on the regional concentration of Xe. Data are mean ± SE in 6 sensitised and challenged rabbits and 3 controls. *: p < 0.05 vs. outside of VD, #: p < 0.05 vs. control. |
The bronchial tree is a branching structure leading to terminal acini (clusters of alveoli) where gas exchange occurs. Previously, computational modelling studies have shown that due to feedback between regional tidal expansion and airway constriction, even a uniform constriction of airway smooth muscle above a critical level can lead to self-organised emergence of patchy areas of ventilation defects resulting from clustered peripheral airway narrowing and closure [3]. This led to the theory that the appearance of ventilation defects may emerge from the complex dynamic system behaviour of the lung.
Our findings further the understanding of uneven ventilation and show that in asthma, airway inflammation is locally heterogeneous, and that this phenomenon is involved in determining the heterogeneity of regional airway constriction and that of regional ventilation following exposure to an allergen. Moreover, the global response to an allergen that causes central airway narrowing is strongly correlated to airway wall infiltration by inflammatory cells such as eosinophils. These findings bring significant new insight to the current understanding of the emergence of heterogeneous airway constriction, and have important implications for the treatment strategies for allergic asthma.
Principal publication and authors
S. Layachi (a), L. Porra (b,c), G. Albu (d), N. Trouillet (e), H. Suhonen (c), F. Peták (f), H. Sevestre (e), P. Suortti (b), A. Sovijärvi (g), W. Habre (d) and S. Bayat (a), J Appl Physiol 115, 1057-1064 (2013).
(a) Université de Picardie Jules Verne and Amiens University Hospital (France)
(b) Department of Physics, University of Helsinki (Finland)
(c) ESRF
(d) Geneva Children’s Hospital, University Hospitals of Geneva, and Geneva University (Switzerland)
(e) Department of Pathology, Amiens University Hospital (France)
(f) Department of Medical Physics and Informatics, University of Szeged (Hungary)
(g) Department of Clinical Physiology and Nuclear Medicine, Helsinki University Central Hospital (Finland)
References
[1] A.R. Sovijarvi, L. Poyhonen, L. Kellomaki and A. Muittari, Thorax 37, 516-520 (1982).
[2] S. Bayat, L. Porra, G. Albu, S. Layachi, F. Petak, P. Suortti, Z. Hantos, A.R.A. Sovijarvi and W. Habre, Am J Respir Crit Care Med 181, A5029 (2010).
[3] J.G. Venegas, T. Winkler, G. Musch, M.F. Vidal Melo, D. Layfield, N. Tgavalekos, A.J. Fischman, R.J. Callahan, G. Bellani, and R.S. Harris. Nature 434, 777-782 (2005).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.