- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2010
- Structural biology
- How a new class of antibiotic inhibits bacterial type IIA toposiomerases
How a new class of antibiotic inhibits bacterial type IIA toposiomerases
The increase in antibiotic resistance and the lack of new therapies in development is a cause of medical, public, and governmental concern. The EU and US have established a Transatlantic Task Force to address antimicrobial resistance, and the Infectious Diseases Society of America has announced a call to action for a global commitment to develop 10 novel antibacterial drugs by 2020 [1].
Bacteria have two type IIA topoisomerases, DNA gyrase and topoisomerase IV. These enzymes are challenging targets for crystallography because they are highly flexible molecular machines. Type IIA topoisomerases regulate DNA topology by: (i) creating a four base-pair staggered double-stranded break in one DNA duplex, (ii) passing another DNA duplex through this break, and (iii) resealing the break. Quinolone antibiotics stabilise double-stranded breaks in DNA, killing bacteria. Quinolone antibiotics have been used in the clinic for over 40 years, but resistance has developed. Target mediated resistance to quinolones most frequently occurs through mutation of two residues in the GyrA subunits of DNA Gyrase (and the equivalent residues in the ParC subunit of topoisomerase IV). Dual targeting of the two enzymes by the antibiotic decreases the chance of the development of resistance.
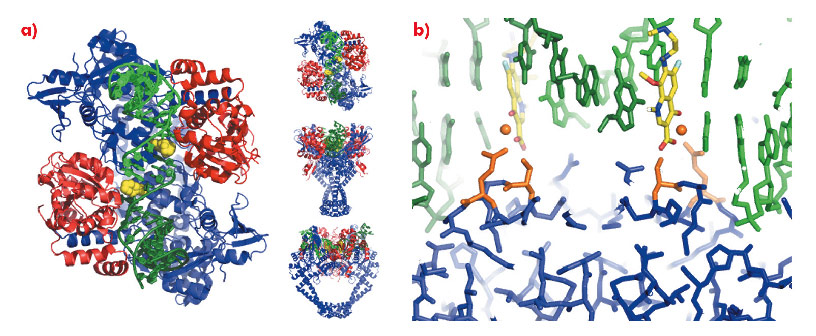
To understand the molecular basis of quinolone action and to investigate new ways of exploiting these clinically validated antibacterial targets [2] we solved the 3.25 Å crystal structure of moxifloxacin in complex with A. baumannii topoisomerase IV and DNA [3], using data collected at the ESRF. As expected the structure showed two quinolone molecules binding four base-pairs apart in the cleaved DNA (Figure 109a), with the slightly wedge-shaped quinolone seeming to exploit the natural tendency of the enzyme to bend the DNA at the cleavage sites (Figure 109b). A magnesium ion, coordinated by two oxygens from moxifloxacin and four water molecules, was seen mediating interactions between the compound and the protein (Figure 109c). Two of the water molecules coordinating the magnesium ion were observed to make hydrogen bonds with the side-chains of residues Ser 84 and Glu 88, the two residues most frequently mutated in quinolone resistant bacteria.
 |
|
Fig. 109: a) The 3.25 Å crystal structure of A. baumannii topoisomerase IV (ParC subunits blue, Par E subunits red), DNA (green) and moxifloxacin (yellow). b) Ser 84 and Glu 88 and the moxifloxacin associated Mg ion(s) are in orange. |
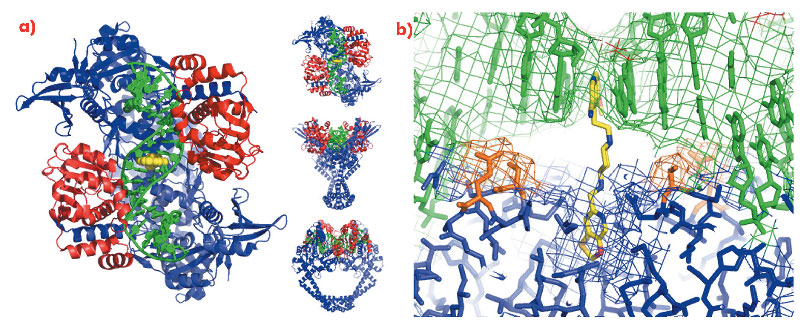
A 2.1 Å crystal structure of a novel bacterial topoisomerase inhibitor (NBTI) in complex with DNA and S. aureus DNA gyrase [4], showed the compound sitting on the twofold axis of the complex with one end buried in a pocket between the two GyrA subunits and the other end sitting between the central two base-pairs of the un-cleaved DNA (Figure 110). Consistent with the good activity of NBTIs against quinolone resistant strains of bacteria, the NBTI did not interact with either Ser 84 or Glu 88. The NBTI binding site is distinct from the quinolone binding site and the mode of action of NBTIs is different from quinolones [4]. The unprecedentedly high-resolution structure for a complex of a type IIA topoisomerase with DNA also gave new insights into the cleavage mechanism of type IIA topoisomerases [4].
 |
|
Fig. 110: a) The 2.1 Å crystal structure of S. aureus DNA gyrase (GyrA subunits blue, GyrB subunits red), DNA (green) and GSK299423 (yellow). b) Ser 84 and Glu 88 are highlighted in orange. |
The collection of a 2.1 Å dataset at the ESRF on a weakly diffracting crystal with a 93 x 93 x 412 Å cell led to the determination of a crystal structure which has provided a structural platform to help with the development of a new class of antibacterial agents.
Authors
B.D. Bax (a), P.F. Chan (b), D.S. Eggleston (c), A. Fosberry (c), D.R. Gentry (b), F. Gorrec (c), I. Giordano (a), M.M. Hann (a), A. Hennessy (a), M. Hibbs (c), J. Huang (b), E. Jones (c), J. Jones (c), K.K. Brown (b), C.J. Lewis (c), E.W. May (b), M.R. Saunders (a), O. Singh (a), C.E. Spitzfaden (a), C. Shen (b), A. Shillings (c), A.J. Theobald (c), A. Wohlkonig (a), N.D. Pearson (b) and M.N. Gwynn (b).
(a) GlaxoSmithKline, Stevenage (UK)
(b) GlaxoSmithKline, Collegeville, Pennsylvania (USA)
(c) GlaxoSmithKline, Harlow (UK)
References
[1] http://www.idsociety.org/Content.aspx?id=4810
[2] D.J. Payne, M.N. Gwynn, D.J. Holmes and D.L. Pompliano, Nature Rev. Drug Discovery 6, 29-40 (2007).
[3] A. Wohlkonig, P.F. Chan, P.A. Fosberry, P. Homes, J. Huang, M. Kranz, V.R. Leydon, T.J. Miles, N.D. Pearson, R.L. Perera, A.J. Shillings, M.N. Gwynn and B.D. Bax, Nature. Struc. Mol. Biol 17, 1152-1153 (2010).
[4] B.D. Bax, P.F. Chan, D.S. Eggleston, A. Fosberry, D.R. Gentry, F. Gorrec, I. Giordano, M.M. Hann, A. Hennessy, M. Hibbs, J. Huang, E. Jones, J. Jones, K.K. Brown, C.J. Lewis, E.W. May, M.R. Saunders, O. Singh, C.E. Spitzfaden, C. Shen, A. Shillings, A.J. Theobald, A. Wohlkonig, N.D. Pearson and M.N. Gwynn, Nature 466, 935-940 (2010).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.