- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2007
- Structural Biology
- Molecular mechanisms of oncogenic mutations in the phosphoinositide 3-kinase catalytic subunit
Molecular mechanisms of oncogenic mutations in the phosphoinositide 3-kinase catalytic subunit
Phosphoinositide 3-kinases (PI3Ks) and their lipid product, phosphatidylinositol-(3,4,5)-trisphosphate (PtdIns(3,4,5)P3), play key roles in many cellular processes [1,2]. Aberrations in PtdIns(3,4,5)P3 levels, either through activation of PI3Ks or through inactivation of the lipid phosphatase PTEN, occur frequently in numerous forms of cancers. For example, recent data suggest that at least 50% of human breast cancers involve mutations in either PI3K![]() or PTEN [3,4]. Cancer-causing mutations increase the activity of PI3K
or PTEN [3,4]. Cancer-causing mutations increase the activity of PI3K![]() . This enzyme is composed of a 110 kDa catalytic subunit (p110
. This enzyme is composed of a 110 kDa catalytic subunit (p110![]() ) and a tightly-associated regulatory subunit (p85). The effect of the regulatory subunit binding to the catalytic subunit is to stabilise the latter and to decrease its basal activity. The catalytic and regulatory subunits interact via the N-terminal adaptor-binding domain (ABD) of the p110
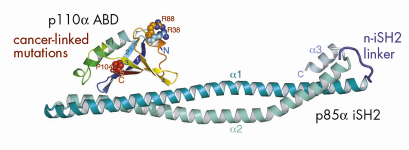
) and a tightly-associated regulatory subunit (p85). The effect of the regulatory subunit binding to the catalytic subunit is to stabilise the latter and to decrease its basal activity. The catalytic and regulatory subunits interact via the N-terminal adaptor-binding domain (ABD) of the p110![]() subunit and the coiled-coil region between the two SH2 domains of the regulatory subunit, i.e., the iSH2 domain. Our crystallographic work carried out at beamline ID14-4 resulted in the structure of a complex of the ABD with the iSH2 domain (Figure 63). Based on this structure and a model of the catalytic core of p110
subunit and the coiled-coil region between the two SH2 domains of the regulatory subunit, i.e., the iSH2 domain. Our crystallographic work carried out at beamline ID14-4 resulted in the structure of a complex of the ABD with the iSH2 domain (Figure 63). Based on this structure and a model of the catalytic core of p110![]() derived from our earlier work at the ESRF [5], we devised a model of the minimum regulated p110/p85 heterodimer. We find that cancer-causing mutations in the ABD are not at the interface of the ABD and the iSH2 domain, and our model of the intact heterodimer suggests that such mutations may affect how the ABD interacts with the remainder of the p110 catalytic subunit.
derived from our earlier work at the ESRF [5], we devised a model of the minimum regulated p110/p85 heterodimer. We find that cancer-causing mutations in the ABD are not at the interface of the ABD and the iSH2 domain, and our model of the intact heterodimer suggests that such mutations may affect how the ABD interacts with the remainder of the p110 catalytic subunit.
 |
|
Fig. 63: A Ribbon representation of ABD/iSH2 heterodimer. The three residues in the ubiquitin-like ABD that were identified as sites of somatic mutations in colon cancers (shown in spheres) are not at the interface of the ABD with the coiled-coil iSH2 domain. |
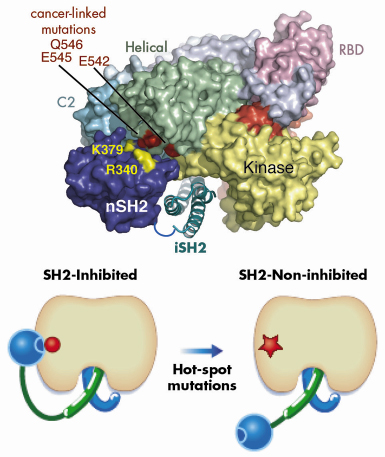
Our model also suggested a mechanism for the functioning of a second type of cancer-causing mutation in p110![]() . Hot-spot cancer-causing mutations in the helical domain of the p110 catalytic subunit eliminate negative charge in a small region on the surface of the helical domain. Based on our model of the heterodimer, we proposed that in the wild type enzyme this negative patch interacts with positive charges on the surface of the N-terminal of the two SH2 domains (nSH2) in the regulatory subunit and this results in inhibition of the basal activity of the catalytic subunit (Figure 64). Eliminating negative charges in this area of the helical domain would lessen the interactions between the two domains and result in a loss of inhibition of the catalytic subunit by the regulating subunit. We tested this hypothesis by constructing a regulatory subunit in which we swapped key positive charges for negative ones. We found that this mutant regulatory subunit inhibited the oncogenic form of the catalytic subunit, but not the wild type form of the catalytic subunit. These studies extend our understanding of the architecture of PI3Ks and provide insight into how two classes of mutations that cause a gain in function can lead to cancer.
. Hot-spot cancer-causing mutations in the helical domain of the p110 catalytic subunit eliminate negative charge in a small region on the surface of the helical domain. Based on our model of the heterodimer, we proposed that in the wild type enzyme this negative patch interacts with positive charges on the surface of the N-terminal of the two SH2 domains (nSH2) in the regulatory subunit and this results in inhibition of the basal activity of the catalytic subunit (Figure 64). Eliminating negative charges in this area of the helical domain would lessen the interactions between the two domains and result in a loss of inhibition of the catalytic subunit by the regulating subunit. We tested this hypothesis by constructing a regulatory subunit in which we swapped key positive charges for negative ones. We found that this mutant regulatory subunit inhibited the oncogenic form of the catalytic subunit, but not the wild type form of the catalytic subunit. These studies extend our understanding of the architecture of PI3Ks and provide insight into how two classes of mutations that cause a gain in function can lead to cancer.
 |
|
Fig. 64: A model illustrating the inhibitory contact between the nSH2 domain and the helical domain of p110 |
Acknowlegements
We thank M. Waterfield and B. Vanhaesebroeck for the p110a plasmid, A. McCarthy for help at ESRF beamline ID14-4, and M. Girvin and G. Gerfen for helpful discussions. The work was supported by AstraZeneca (RLW), MRC (RLW) and NIH GM55692 (JMB).
References
[1] L.C. Cantley, Science 296, 1655 (2002).
[2] M.P. Wymann, M. Zvelebil, M. Laffargue, Trends Pharmacol Sci 24, 366 (2003).
[3] J. Luo, B.D. Manning, L.C. Cantley, Cancer Cell 4, 257 (2003).
[4] L.H. Saal, K. Holm, M. Maurer, L. Memeo, T. Su, X. Wang, J.S. Yu, P-O. Malmström, M. Mansukhani, J. Enoksson, H. Hibshoosh, Å. Borg, R. Parsons, Cancer Res. 65, 2554 (2005).
[5] E.H. Walker, O. Perisic, C. Ried, L. Stephens, R.L. Williams, Nature 402, 313 (1999).
Principal publication and authors
N. Miled (a,b), Y. Yan (c), W-C. Hon (a), O. Perisic (a), M. Zvelebil (d), Y. Inbar (e), D. Schneidman-Duhovny (e), H.J. Wolfson (e), J.M. Backer (c) and R.L. Williams (a), Science 317, 239 (2007).
(a) MRC Laboratory of Molecular Biology, Cambridge (UK)
(b) Laboratoire de Biochimie et de Génie Enzymatique des Lipases, Ecole Nationale d’Ingenieurs Sfax (Tunisia)
(c) Albert Einstein College of Medicine, NY (USA)
(d) Ludwig Institute for Cancer Research, London (UK)
(e) School of Computer Science, Tel Aviv University (Israel)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.