- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Enabling technologies
- Fully automatic data collection: a new tool for structural biologists
Fully automatic data collection: a new tool for structural biologists
Protein crystallography is essential in the understanding of fundamental biological processes and the design of new drugs. The process involves characterising hundreds of crystals and collecting optimised data sets, a laborious task performed non-stop day and night. A fully automated system that will mount, screen and collect the best possible data from hundreds of crystals without human intervention, 24 hours a day, has now been developed.
Considerable human effort is dedicated to evaluating protein crystals at synchrotron sources in order to find the few crystals that diffract well or to find the one crystal that contains a potential new drug candidate. Much of this work is repetitive and the time spent could be better invested in the interpretation of results. MASSIF-1 is a unique facility for the high throughput, fully automatic characterisation and data collection of crystals of macromolecules [1]. The combination of ESRF developed robotic sample handling (RoboDiff) and advanced software protocols [2] has provided a new tool to structural biologists. The beamline is not designed to replace user visits to the synchrotron but rather to do the hard work of screening crystals or collecting data sets through the night, freeing researchers to spend time on more challenging data collection problems and study the underlying biology.
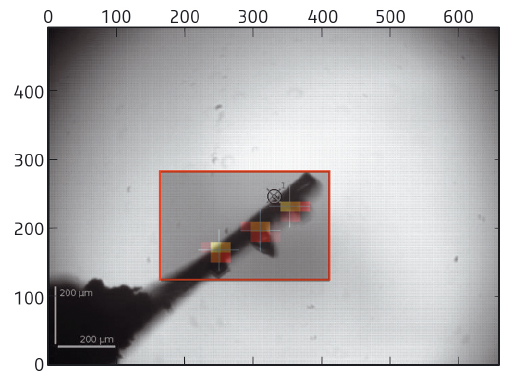
At the core of the beamline is a fully automatic system to mount, locate, centre to the optimal diffraction volume, characterise and collect data, if possible, from multiple cryocooled crystals. Using the capabilities of pixel array detectors, the system operates at about the same speed as a human operator, taking an average of 6 minutes per sample, but performing far more than is possible with a user present, without getting tired. In the first step, the sample mount is analysed in order to determine the area to be scanned (Figure 152). A fast X-ray based routine is then launched to locate crystals and centre them to the beam systematically at the position of highest diffraction signal. The routine is often able to locate crystals more effectively than the human eye, either as they are mounted in opaque medium or a large excess of liquid causes lens effects. In many cases, diffraction data have been obtained when centring a crystal was not possible manually. Once centred, the samples are characterised (assessed for quality, diffraction limit, crystallographic symmetry) and important parameters, such as flux, beam size and crystal volume, are automatically taken into account, ensuring calculation of optimal data collection strategies. Optimised data sets are then collected and automatically processed [3] before the next sample is mounted.
 |
|
Fig. 152: Sample mounts are analysed and an area chosen to scan through the X-ray beam (red box). Analysis of the diffraction images obtained yields a heat map defining the location and diffraction quality of crystals. Optimum points are then selected for data collection (crosses), in this case, three separate crystals. |
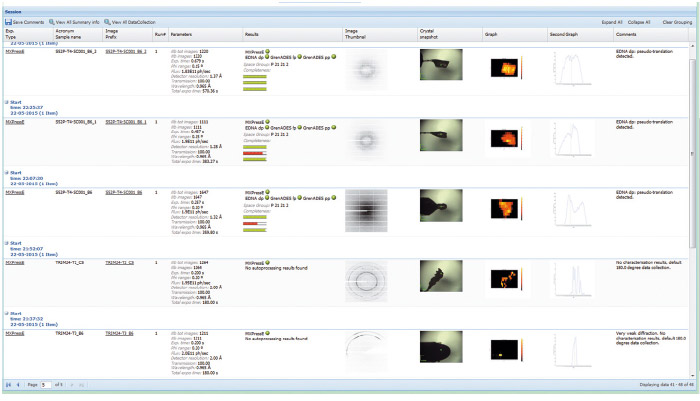
Automation necessarily involves standardisation; however, for an automatic data collection service to be successful for a range of projects, considerable flexibility in data collection parameters must be allowed. This has been implemented by allowing user preferences (such as type of data collection, resolution required, beam size, etc.) to be entered at the sample level in the beamline database ISPyB [4] that also displays the results for each sample (Figure 153). An online booking system adds to the flexibility: rather than the rigid system in place at other beamlines, users can book time on the MASSIF-1 calendar with very short notice and samples then enter a queuing system.
 |
|
Fig. 153: A screenshot showing the display in ISPyB of results, such as diffraction maps and autoprocessing, for a series of samples processed at MASSIF-1. Comments are automatically written (far right column) to inform users on various stages of the process such as ‘weak diffraction’, default 180° data collection’, etc. |
Over 15,000 samples have now been processed with no human intervention allowing the accumulation and comparison of a large amount of information that was previously unknown, including the exact dimensions of crystals and further information about their quality. This has been fed back into the software in order to improve data collection. For example, the most commonly observed crystal length is 50 µm – this led to the selection of a default beam size that best reflects the samples. The latest developments now include options to allow the collection of data sets from multiple crystals on the same sample support (Figure 152) and more advanced data collection strategies such as helical data collection.
The new level of automation should decrease project lifecycles and, in partnership with developments being made in the automatic mounting of crystals [5], a fully automatic pipeline from protein to structure can now be envisioned.
Principal publication and authors
Fully automatic characterization and data collection from crystals of biological macromolecules, O. Svensson (a), S. Monaco (a), A.N. Popov (a), D. Nurizzo (a) and M.W. Bowler (b), Acta Cryst. D 71, 1757-1767 (2015); doi: 10.1107/S1399004715011918.
(a) ESRF
(b) European Molecular Biology Laboratory, Grenoble Outstation (France)
References
[1] M.W. Bowler et al., J. Synch. Rad. 22, 1540-1547 (2015).
[2] S. Brockhauser et al. Acta Cryst. D 68, 975-984 (2012).
[3] S. Monaco et al. J. App. Cryst. 46, 804-810 (2013).
[4] S. Delagenière et al. Bioinformatics 27, 3186-3192 (2011).
[5] F. Cipriani et al. Acta Cryst. D 68, 1393-1399 (2012).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.