- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2003
- Materials Science
- Solving Larger Molecular Crystal Structures from High-resolution Powder Diffraction Data
Solving Larger Molecular Crystal Structures from High-resolution Powder Diffraction Data
Powder diffraction is an important and often unique crystallographic tool to determine the structures of polycrystalline materials for which suitable single crystals cannot be grown. This includes, for example, certain crystalline forms of pharmaceutically-active molecules. However, overlap of the Bragg reflections in the powder diffraction pattern obscures the data and reduces the accuracy of the extracted diffraction-peak intensities. Consequently, traditional single-crystal methods for crystal structure determination are less reliable and often fail when used with powder data, limiting the number of atoms that can be located by this approach to around 30 for equal-atom structures such as organic molecules.
Significant improvement in data quality can now be obtained. Herein, we illustrate this with the routine direct-methods solution of a 48-atom fully-organic crystal structure, which was achieved by exploiting anisotropic thermal expansion, using high-resolution powder X-ray diffraction.
One way to reduce the effective peak overlap is to exploit the anisotropy in thermal expansion that many low-symmetry materials naturally exhibit [1]. At different temperatures the relative positions of Bragg reflections change, owing to the anisotropic changes in the lattice dimensions, but maintain approximately the same intensities. By collecting data at multiple temperatures it is possible to assess the individual contribution of reflections that are overlapping at one temperature, but are better resolved at another.
Changes in the overlap of the reflections as the temperature is varied are best revealed with an intrinsically high-resolution instrument, so that the effects of small peak shifts are more readily discernable. The high-resolution powder diffraction beamline has recently been upgraded, by being moved from a bending-magnet (BM16) to an undulator source (ID31), where it receives a significantly more intense incident X-ray beam. The performance is now such that the systematic use of anisotropic thermal expansion to study more-complex crystal structures by powder diffraction can be envisaged.
During our continuing studies on the low temperature structures of small globular organic molecules, we were unable to solve the arrangement of molecules in 9-ethylbicylco[3.3.1]nona-9-ol (C11H20O) despite having deduced the unit cell dimensions, the number of independent molecules in the cell and the crystal symmetry, from measurements made on BM16. Having 48 non-hydrogen atoms in the asymmetric unit (four independent molecules), this complex structure was clearly a promising candidate for an attempt at using the anisotropic thermal expansion approach.
 |
|
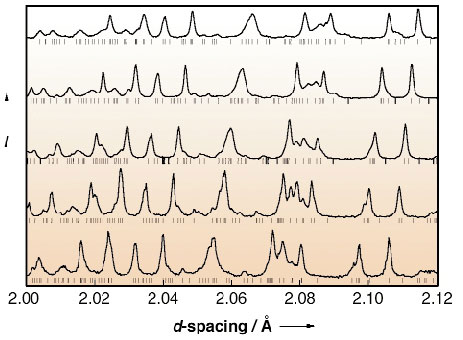
Fig. 36: Selected regions of the five diffraction patterns showing anisotropic thermal expansion. The relative thermal expansion of the a, b and c cell parameters must be different in order to change the relative positions of the peaks. The five diffraction patterns are fitted simultaneously with a single set of peak intensities. Vertical bars indicate Bragg peak positions. |
Five powder diffraction patterns at different temperatures in the range 80 180 K were collected on ID31. Figure 36 illustrates the changes in the diffraction pattern due to peak shifts as a consequence of the anisotropic thermal expansion manifested between 80 K and 180 K. Individual integrated peak intensities were extracted by a multipattern Pawley-style refinement procedure1, fitting a single set of diffraction peak intensities to the multiple data sets. The four independent molecules, in their entirety (Figure 37), could be found simply using the standard single-crystal direct-methods program shelxs plus Fourier recycling.
 |
|
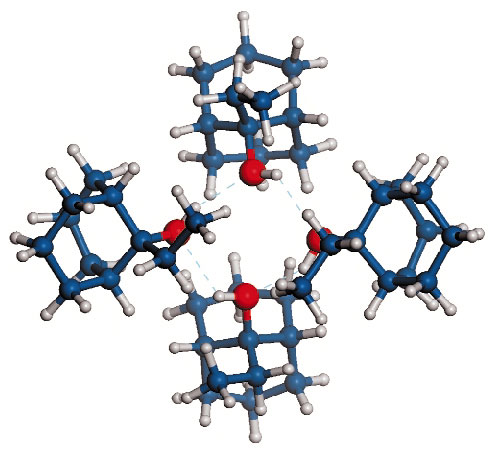
Fig. 37: View of the arrangement of the four independent 9-ethylbicyclo[3.3.1]nona-9-ol molecules. The molecules form a tetramer linked by an approximate square of O-H···O hydrogen bonds between hydroxide groups at the heart of the cluster. The cluster's outer surface is composed of hydrogen atoms. Interactions between adjacent clusters are therefore of a Van-der-Waals nature. |
The solution of this structure illustrates a considerable advance in the complexity of a crystal structure derived by direct methods from powder data, and in the quality of data. The data collection was completed in about 12 hours during the first day of scheduled user experiments on the newly-commissioned beamline, with the structural model being derived the very next day. The speed and simplicity of both the data collection and analysis strongly suggest that even larger structures will also be tractable with high-resolution powder X-ray diffraction data.
References
[1] K. Shankland, W.I.F. David, D.S. Sivia, J. Mater. Chem. 7, 569 (1997).
Principal Publication and Authors
M. Brunelli (a), J.P. Wright (a), G.B.M. Vaughan (a), A.N. Fitch (a) and A.J. Mora (b), Angew. Chem. Int. Ed., 42, 2029-2032 (2003).
(a) ESRF
(b) Universidad de Los Andes, Mérida (Venezuela)
1 Profile matching procedure in which the diffraction profile is calculated as a sum of overlapping reflections and the intensities are variables in a least-squares procedure.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.