- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2010
- Electronic structure and magnetism
- Hydration structure of the bromide ion in aqueous solution
Hydration structure of the bromide ion in aqueous solution
The solvation properties of ions and the role of the surrounding water molecules are important throughout biology and chemistry. While much experimental and theoretical work has been devoted to the characterisation of the structural and dynamical behaviour of cations in water, the hydration properties of halides are still the subject of intense debate. For the bromide ion, the hydration number as determined by X-ray and neutron diffraction and X-ray absorption is in the range 6-7.4 and the Br-O first shell average bond length determined by X-ray diffraction is between 3.19 and 3.40 Å. These scattered results underline the difficulty of defining the halide coordination shells also as a consequence of the fast water exchange between the first and second hydration shells.
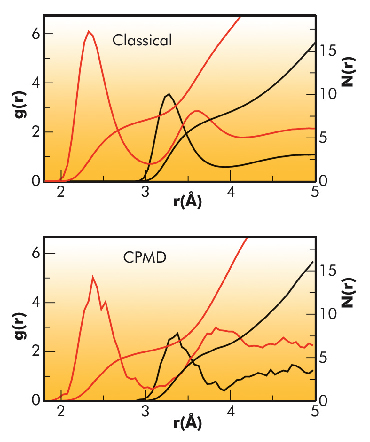
Here we report on the first study of the hydration structure of the bromide aqua ion using a combination of state-of-the-art density functional theory (DFT)-based Car-Parrinello molecular dynamics (CPMD) simulations and extended X-ray absorption fine structure (EXAFS) spectroscopy. This experimental technique is particularly well suited for the investigation of the local solvent structure of ions dissolved in water due to its atomic selectivity and its sensitivity to dilute solutions. Beside the quantum mechanical CPMD simulation, we have also performed a classical molecular dynamics (MD) simulation of the bromide ion in aqueous solution and compared the results obtained from these two different theoretical approaches. Structural arrangements of water molecules around the bromide ion are characterised by the Br-O and Br-H radial distribution functions, and the results obtained from the classical MD and CPMD simulations are depicted in Figure 86. In both cases, the presence of a nonzero first minimum in the Br-O g(r)’s and of a second minimum in the Br-H g(r)’s indicates that the first solvation shell is not well-defined and that several exchange events may take place between the first and second hydration sphere. The first difference between the classical and the CPMD simulation is the Br-water first shell distance, which is shorter in the classical trajectory (RBr-O is 3.33 and 3.27 Å for the CPMD and classical MD simulation, respectively). The picture that emerges from our simulations is that the pair potentials used in the classical approach are too rigid giving rise to a first hydration shell which is too tightly bound to the bromide ion. Conversely, in the CPMD trajectory the first coordination shell is more flexible and unstructured, without a well-defined configuration of water molecules around the ion. Both in the MD and CPMD trajectories, the bromide ion transits among several coordination numbers, but while in the classical case the dominant species existing in solution are seven- and eight-fold hydration complexes, only six-fold complexes play a dominant role in the CPMD case. Moreover, in both simulations the first hydration shell is slightly asymmetric, but the asymmetry is more pronounced in the CPMD trajectory; this asymmetry is responsible for the presence of an induced net dipole moment on the bromide ion (µ = 0.85D), indicating that the bromide electron density is strongly influenced by the presence of the surrounding water molecules and that the inclusion of polarisation effects is essential to provide a correct description of the anion-water interactions.
 |
|
Fig. 86: Br-O (black line) and Br-H (red line) radial distribution functions obtained from the classical MD and CPMD simulations. Running integration numbers are also shown. |
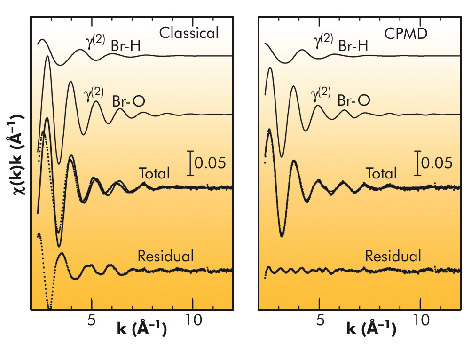
Finally, the MD structural results were compared with EXAFS experimental data. χ(k) theoretical signals have been calculated starting from the classical and CPMD Br-H and Br-O g(r)’s and the structural parameters derived from the simulations were kept fixed during the EXAFS analysis. The importance of the hydrogen contribution to the EXAFS spectra of metal cations in aqueous solution has been previously pointed out and here for the first time it has been possible to also include the hydrogen scattering in the EXAFS analysis of anion aqueous solutions. In the case of the classical MD simulation, a rather poor agreement between the theoretical and experimental spectrum was found, while the spectrum calculated from the CPMD g(r)’s matched the experimental data very well, thus showing that the structural and dynamical information derived from the CPMD simulation was basically correct (Figure 87).
 |
|
Fig. 87: Comparison between the EXAFS theoretical signals (solid line) calculated from the classical MD and CPMD Br-O and Br-H g(r)’s and experimental data (dotted line). The residual signals are also shown. |
In conclusion, we have shown that advanced DFT based simulation techniques, which take into account both polarisation effects and many body interactions, are necessary to provide a proper description of the hydration properties of halide anions. Moreover, by taking advantage of the reliable information on the bromide ion local hydration structure obtained from the CPMD trajectory, it has been possible to include the hydrogen scattering in the EXAFS analysis of anion aqueous solutions.
Principal publication and authors
P. D’Angelo (a), V. Migliorati (a) and L. Guidoni (b), Inorg. Chem. 49, 4224 (2010).
(a) Dipartimento di Chimica, Università di Roma “La Sapienza” (Italy)
(b) Dipartimento di Chimica, Ingegneria Chimica e Materiali, Università degli Studi dell’Aquila (Italy)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.