- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Electronic structure and magnetism
- Surface and bulk approach to time-resolved characterisation of heterogeneous catalysis
Surface and bulk approach to time-resolved characterisation of heterogeneous catalysis
Chemical phenomena of both fundamental and technological importance are often very complex in nature. Catalysis is a discipline in which such complexity is paradigmatic. Typically, heterogeneous catalytic processes involve the contact of gases or liquids with nanosized active components. To shed light on these complex systems, multi-technique approaches, often having high time-resolution as well the capability to address surface and bulk properties, appear as optimal tools for unravelling the elusive details of catalysts function. Surface and bulk sensitivity is generally achieved by a combination of at least two techniques such as X-ray and vibrational spectroscopies. Here we highlight work carried out in two fields: the preparation and the evolution under reaction conditions of catalytic materials.
 |
|
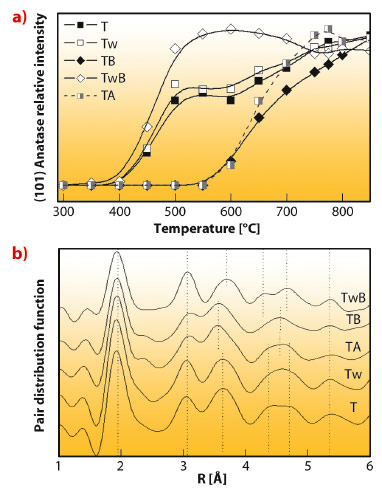
Fig. 104: Room temperature PDF-XRD and XRD peak intensity during a calcination ramp of a series of anatase solid precursors. |
As a first representative example we focus on the morphological control of the anatase polymorph of titanium oxide. Figure 104 displays the structural differences observed using total scattering (PDF-XRD; atom pair density function) among a series of initial, amorphous precursors of titania samples prepared by microemulsion. The figure also includes the corresponding XRD observation of the nucleation and growth process during thermal treatment. The latter is based upon the analysis of the intensity of Bragg diffraction peaks that eventually appear when sufficient crystallinity arises. The initial precursors can pertain to two different groups; initial precursors T, Tw, and TwB (differing in initial synthesis conditions) display rather similar patterns and appear to show significant differences (Figure 104a) with samples TA and TB. Such differences are directly linked to the temperature of nucleation, which divides the materials in the two groups mentioned in relation to their initial precursor structure. This observation provides strong evidence of a thermodynamic control of the anatase nucleation process. Primary particle size is obviously affected by both nucleation and growth, but additional studies demonstrated the key importance of the nucleation. Therefore, an inherently thermodynamic factor was shown to control the size of the (crystalline) anatase phase. This fundamental understanding opens up the possibility for tuning the morphology of nano-anatase by manipulating the initial structure of the precursor materials. More importantly, by a joint analysis of XRD-PDF and infrared spectroscopy, the physical basis for controlling the size and shape of the oxide can be unravelled.
 |
|
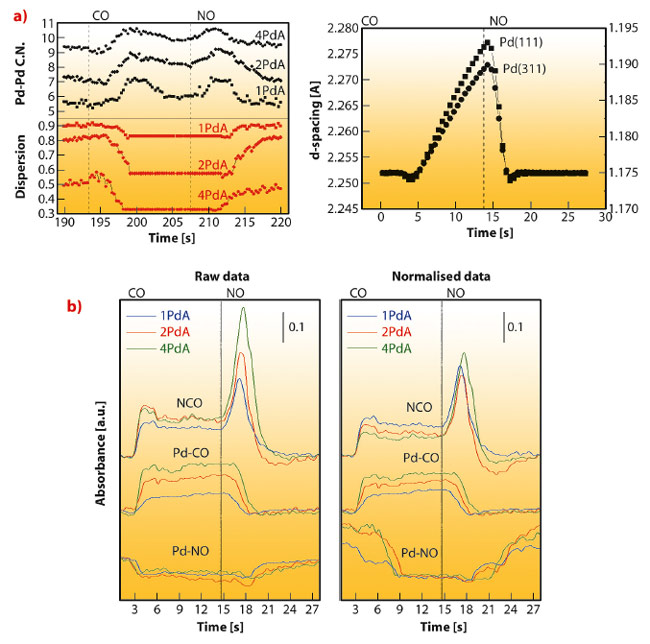
Fig. 105: a) Pd-Pd EXAFS 1st shell coordination number and dispersion for several (1, 2, 4 wt.%) Pd-Al2O3 catalysts and variation of two Pd metal XRD lattice planes for a 2 wt.% Pd-Al2O3 catalyst in a CO-NO atmosphere. b) IR intensity (raw and normalised per Pd surface atom) of characteristic absorbed species behaviour in a CO-NO atmosphere for several (1, 2, 4 wt.%) Pd-Al2O3 catalysts. |
As a second example, Figure 105 summarises the rich chemistry displayed by Pd nanoparticles observed using a CO-NO gas mixture. Figure 105a display the Pd metal phase behaviour through the Pd-Pd EXAFS coordination number (C.N.) and dispersion of the metal phases for a series of catalysts with increasing quantity of palladium. The first result is that the metal responds actively to the gas mixture nature in all cases, showing that the apparent C.N. increases under CO and diminishes -in a fully reversibly manner- to the initial value under NO. This behaviour is quantified in the dispersion plot assuming spherical particles and an exclusive size effect. In Figure 105 we can also observe that we detected a slowly varying region in the C.N. behaviour toward the middle-end part of the CO treatment. This region can be understood with the help of high energy XRD which clearly reveals this transient phenomenon as a saw-tooth variation in the d spacing of the Bragg reflection from Pd nanoparticles (Figure 105a); this in turn is related to the somewhat unexpected formation of a palladium carbide phase, not previously detected under stoichiometric CO-NO mixtures. The synchronous infrared panels of Figure 105b indicate that such PdCx phase is formed from dissociated CO and leads to storage of carbon species at the metal; concomitantly the partitioning of the adsorbed CO between linear and bridged sites is changed with a promotion of the linear bound CO over the bridged. Moreover, the carbide phase is completely removed under NO and a large amount of NCO is formed simultaneously. The reversible chemistry summarised in Figure 105 is connected to two key steps, in turn related to, firstly, surface CO coverage and dissociation at the surface of the Pd and, secondly, the subsequent stripping of C from the metal bulk. Such steps are synchronised with (reversible) size/shape changes of Pd with profound implications in CO/NO elimination reactions. The uncovering of such physical/chemical phenomena, with significance in practical use of pollutant elimination using Pd-TWC systems, is only possible with the combined in situ use of XRD-XAS-IR spectroscopies.
Principal publication and authors
A. Kubacka (a), A. Iglesias-Juez (a), M. Fernández-García (a), M. Di Michiel (b) and M.A. Newton (b), ChemCatChem 4, 725-737 (2012).
(a) Instituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie 2, Madrid (Spain)
(b) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.