- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Electronic structure, magnetism and dynamics
- RIXS reveals where TiO2 nanoparticles store hot electrons after plasmon-induced electron transfer from Au
RIXS reveals where TiO2 nanoparticles store hot electrons after plasmon-induced electron transfer from Au
The chase for efficient photocatalysts is pivotal for clean energy production and understanding charge generation and storage after light absorption is fundamental for materials optimisation. Adding plasmonic nanoparticles to oxides for catalysis is particularly promising. The modification of the TiO2 electronic structure following charge transfer from Au nanoparticles was studied with resonant inelastic X-ray scattering (RIXS) to address the question of where TiO2 stores the additional charge.
Titanium dioxide is one of the reference materials for photocatalysis. Its characteristics should make it an ideal photocatalyst for water splitting, but it absorbs sunlight poorly because of its wide energy gap [1]. Adding an efficient light absorber able to transfer energy to TiO2 is a means to enhance its photo-catalytic activity, for example in dye-sensitised solar cells. Plasmonic nanoparticles are outstanding light absorbers due to the localised surface plasmon resonance (LSPR). Devices coupling TiO2 and plasmonic materials show promising results, indicating that charge transfer to TiO2 is indeed triggered by LSPR excitation [2]. However, efficient charge transfer is essential but not sufficient for optimal photocatalysts as the catalytic process is slow and requires long-lived charges on active sites. We investigated modifications of the TiO2 electronic structure induced by plasmon-induced long-lived charges and addressed the question of where TiO2 stores the transferred charge.
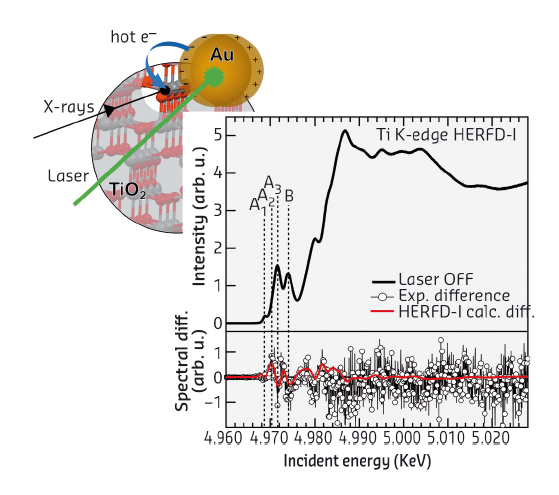
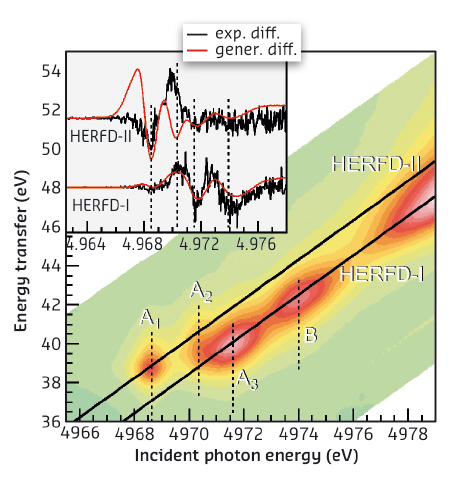
High-energy-resolution fluorescence-detected (HERFD) XANES at the Ti K-edge is obtained by collecting only a small energy bandwidth (~1 eV) at the maximum of the Kβ1,3 line. We excited the LSPR of Au with a continuous laser in a TiO2/Au powder and simultaneously probed Ti electronic structure by HERFD-XANES at the Ti K-edge at beamline ID26. By subtracting laser on and laser off measurements, we obtained a non-zero spectral difference (Figure 54), which is dominated by contributions from excited states with long lifetimes (>μs) as both X-ray probing and laser irradiation were continuous [3]. The detected signal is thus a signature of long-lived charges that affect the electronic structure of a small percentage of Ti atoms. Measurements on bare N-doped TiO2 confirm that Au supplies charges. To obtain more information on how the injected charge is trapped, we recorded full Kβ profiles at incident energies across the pre-edge. The resulting distribution of spectral intensity along the incident and the transferred energy axis is called a RIXS plane with diagonal cuts corresponding to a HERFD-XANES scan (Figure 55). Four characteristic pre-edge peaks, A1, A2, A3 and B, are found on different HERFD diagonals: A3 and B on HERFD-I, and A1 and A2 on HERFD-II. This separation reflects different contributions of Ti atomic orbitals to the spectral features, with contributions from more delocalised orbitals (Ti p) lying on HERFD-I and those from more localised orbitals (Ti d) on HERFD-II.
 |
|
Fig. 54: Schema of the process: laser excitation of LSPR of Au nanoparticles induces hot electron injection and trapping. The graph shows TiO2 HERFD-XANES (top) and experimental and calculated (red-shifted minus unshifted) spectral differences (bottom). |
We also collected the spectral difference induced by LSPR excitation on HERFD-II and found small but marked differences in the pre-edge (inset Figure 55), as for HERFD-I. The HERFD-I spectral difference can be modelled by a –0.8 eV spectral shift, while the difference observed on HERFD-II arises from a modulation of the A1 and A2 intensities. This twofold behaviour provides important insight making it possible to understand that, after charge injection, some photo-induced charges remain trapped in Ti p-orbitals and the signal coming from these sites appears red-shifted compared to the rest of Ti atoms. The modulation of A1 and A2 intensities is inconsistent with a charge trapped in Ti d-orbitals, i.e. with reduction of Ti4+ into Ti3+, but can be assigned to small structural rearrangements needed to accommodate the charge in Ti p-orbitals. Pre-edges are indeed very sensitive to p-d hybridisation.
 |
|
Fig. 55: 1s3p RIXS plane of Ti pre-edge of N-doped TiO2. The characteristic four peaks A1-3 and B lie along two different cuts, HERFD-I (A1 and A2) and HERFD-II (A3 and B). The inset shows the experimental difference (black) between laser on and laser off scans compared to the difference between the spectrum recorded without laser and the same spectrum red-shifted by 0.8 eV (red). A red-shift models the spectral difference observed on HERFD-I, while the effect on HERFD-II is not a shift but a modulation of A1 and A2 intensities. |
In conclusion, following LSPR excitation, charges are injected from Au into TiO2 and the longest lived ones are trapped in Ti p-orbitals. Traps are probably distorted TiO3 octahedra near the Au/TiO2 interface. The possibility to induce trapped charges with long lifetimes at the surface via LSPR excitation endorses the use of plasmonic nanoparticles to improve TiO2 catalytic activity.
Principal publication and authors
Probing long-lived plasmonic-generated charges in TiO2/Au by high-resolution X-ray absorption spectroscopy, L. Amidani (a,b), A. Naldoni (c), M. Malvestuto (d), M. Marelli (c), P. Glatzel (b), V. Dal Santo (c) and F. Boscherini (a), Angewandte Chemie Int. Ed. 54, 5413-5416 (2015); doi: 10.1002/anie.201412030.
(a) Department of Physics and Astronomy, University of Bologna, Bologna (Italy)
(b) ESRF
(c) CNR-Istituto di Scienze e Tecnologie Molecolari, Milano (Italy)
(d) Elettra-Sincrotrone Trieste, Trieste (Italy)
References
[1] A. Fujishima, K. Honda, Nature 238, 37 (1972).
[2] S. Mubeen et al., Nat. Nanotechnol. 8, 247 (2013).
[3] M.H. Rittmann-Frank et al., Angew. Chem. Int. Ed. 53, 5858 (2014).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.