- Home
- News
- Spotlight on Science
- X-ray diffraction...
X-ray diffraction reveals the stabilisation effects of a hyperstable engineered protein catalyst
09-12-2020
X-ray crystal structures solved at beamline ID23-1 of a computationally designed hyperstable enzyme (Tm = 73.5°C) reveal an unprecedented double-lock system that massively reduces volumes of enzyme access tunnels. Tracing of the access tunnels using krypton-derivative crystals demonstrates that transport of ligands is still effective.
Share
An X-ray diffraction (XRD) experiment at beamline ID23-1 uncovered unprecedented molecular details of the inner organisation and workings of a hyperstable engineered biocatalyst. Specifically, a ‘soak-and-freeze’ crystal derivatisation technique [1] made it possible to gain valuable information on the functionality of intra-macromolecular pathways connecting a buried catalytic centre with bulk solvent.
A fundamental role of proteins is to act as enzymes – biocatalysts that increase the rate of almost all chemical reactions within living systems. Enzymes have evolved for billions of years, and will continue to do so as long as life on earth exists. Many of them have been successfully incorporated into diverse industrial, environmental and biomedical technologies. Often, wild-type enzymes do not fully meet the demands of these harsh technological processes, and punctual mutations are engineered into them to improve their physico-chemical properties for technological applications. The key parameter for all enzymes to be employed in industrial catalysis is thermostability, which allows them to withstand elevated temperatures during biocatalytic processes.
Haloalkane dehalogenases (HLDs; EC 3.8.1.5) are α/β-hydrolases that catalyse the hydrolytic cleavage of the carbon–halogen bond in diverse halogenated aliphatic hydrocarbons via SN2 nucleophilic substitution. The reaction requires the addition of a water molecule and releases a halide ion together with a proton, finally producing the corresponding alcohol. Structurally, HLDs consist of a canonical α/β-hydrolase fold, which is composed of a central eight-stranded β-sheet domain surrounded by several α-helices (i.e. αβα sandwich architecture). An additional versatile helical cap domain is observed to be specific to each HLD enzyme. In all HLDs, the active site is positioned in a hydrophobic pocket buried between the α/β-fold core and the cap domain, and this catalytic centre is connected with the bulk solvent via a main tunnel and a slot tunnel. Both of these tunnels are crucial determinants of the specific catalytic activity and the substrate selectivity of each HLD enzyme [2].
Recently, a fully automated and robust computational pipeline, FireProt [3,4], was developed, combining energy- and evolution-based approaches to design highly stable multi-point mutant proteins. FireProt was employed to enhance the thermostability of DhaA, an HLD enzyme from Rhodococcus rhodochrous (Tm = 50.5 °C; Topt = 45 °C). After several iteration cycles, an 11-point DhaA mutant was obtained, hereafter referred to as DhaA115, with outstanding thermostability (Tm = 73.5 °C) and thermophilicity, as demonstrated by a substantial shift in the optimal catalytic temperature (Topt = 65 °C) [1]. An understanding of the structural basis of this hyperstabilisation was required in order to develop next-generation computer algorithms and predictive tools.
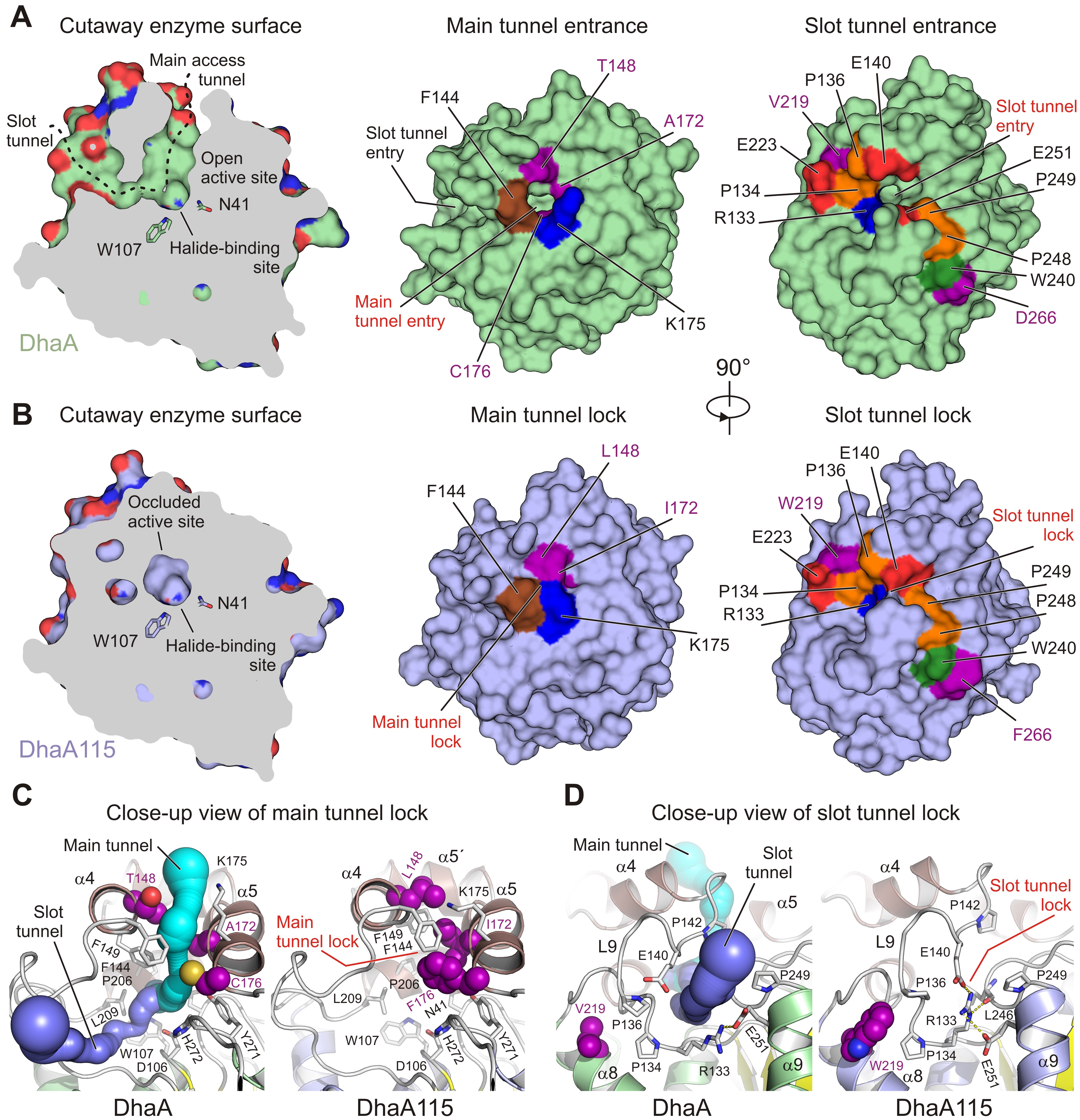
Fig. 1: Visualisation of access tunnels. Cutaway surface representations of enzyme active site and access tunnels for the wild type DhaA (a) and the hyperstable DhaA115 (b) enzymes. The DhaA enzyme has a so-called open active site connected with the outside bulk solvent via both the main and slot access tunnels. In contrast, in DhaA115, the main and slot access tunnels are, to a large extent, blocked because of the direct or indirect effects of stabilising mutations. Visualisation of the CAVER-calculated main (c) and slot (d) access tunnels. The main tunnel was calculated (cyan) for wild-type DhaA (left panel). The tunnel is absent in DhaA115 because of three stabilising residues (L148, I172 and F176) which, together with other hydrophobic and aromatic residues (F144, F149 and L209) form the so-called main tunnel lock (right panel). Similarly, the slot tunnel calculated (blue) for wild type DhaA (left panel) is absent in DhaA115 because of a conformational change of the L9 loop triggered by the presence of stabilising residue W219 and a positional shift of the α9 helix. This new constellation allows the residues R133 and E140 to form a so-called slot tunnel lock (right panel). The stabilising residues are shown as purple spheres, and the other protein residues are shown as grey sticks.
To fill this gap, the hyperstable enzyme DhaA115 was crystallised and high-resolution X-ray structures were solved. Analyses of these crystal structures highlight specific amino acid constellations that primarily reinforce the αβα-sandwich architecture and the helical cap domain via multiple newly established interactions of the non-polar, hydrophobic and aromatic π–π stacking types. Surprisingly, it was found that placement of bulky aromatic amino acids on the protein surface triggered some unexpected long-distance changes in the protein backbone. Essentially, these changes cause the gates and the internal volumes of both the main and the slot access tunnels to be restricted and, consequently, the enzyme active site appears somewhat occluded (Figure 1). Interestingly, despite the active site occlusion, experimental mapping of the enzyme tunnels by krypton derivatisation of the DhaA115 crystals, supported by computational protein biology tools [5,6], showed that ligand molecules can still be transported through the enzyme tunnels (Figure 2).
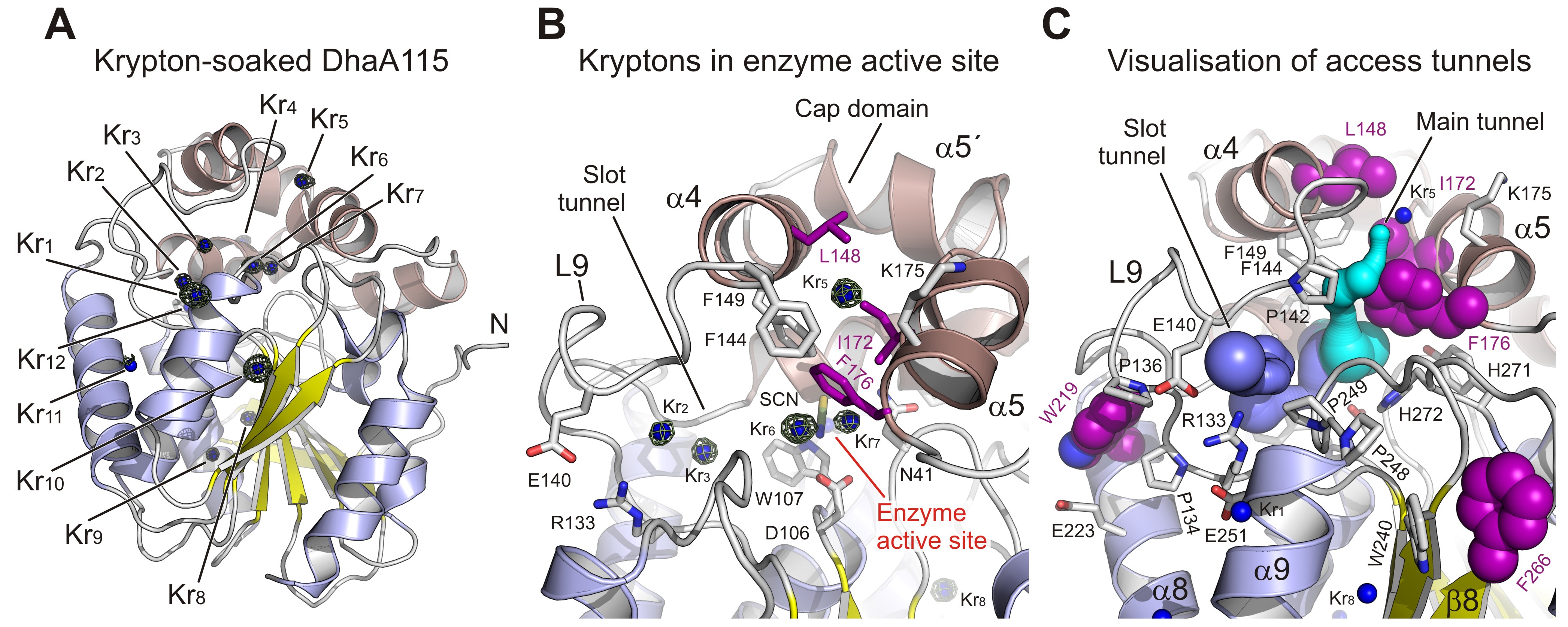
Fig. 2: Experimental tracking of the enzyme access tunnels. a) Cartoon representation of DhaA115 krypton derivative crystal structure with the central eight-stranded β-sheet (yellow), α/β-hydrolase helices (blue) and helical cap domain (brown). The locations of the krypton atom sites (Kr1 to Kr11) in pressurised DhaA115 are shown as blue spheres with anomalous Fourier maps contoured at 4σ (green mesh). b) Close-up view of the active site and access tunnels in the DhaA115 krypton derivative crystal structure. Two krypton atoms (Kr6 and Kr7) are found in the enzyme active sites, two (Kr2 and Kr3) occupy the slot tunnels, and one krypton (Kr5) sits on top of the cap domain. The stabilising residues are shown as purple sticks and the other protein residues are shown as grey sticks. c) Visualisation of the access tunnels in the DhaA115 krypton derivative crystal structure calculation by CAVER. The main (cyan) and slot (blue) tunnels are partially restored upon pressurisation. The main tunnel is depicted in cyan and the slot tunnel in light blue. The stabilising residues are shown as purple spheres, remaining residues are shown as grey sticks, and krypton atoms as blue spheres.
Collectively, these results demonstrate that the hyperstabilisation engineered in DhaA leads to a massive reduction in the volume of its access tunnels, and that the enzymes are still capable of operating since they are permeable to substrates, products and water molecules. This permeability is then increased at elevated temperature, as previously demonstrated by the shifted optimal catalytic temperature (Topt = 65 °C) of the DhaA115 enzyme. These findings highlight key thermostabilisation effects and provide a structural basis for designing new thermostable protein catalysts.
Principal publication and authors
Decoding the intricate network of molecular interactions of a hyperstable engineered biocatalyst, K. Markova (a,b), K. Chmelova (a), S.M. Marques (a,b), P. Carpentier (c,d), D. Bednar (a,b), J. Damborsky (a,b), M. Marek (a,b), Chem. Sci. 11, 11162-11178 (2020); https://doi.org/10.1039/D0SC03367G.
(a) Loschmidt Laboratories, Masaryk University, Brno (Czech Republic)
(b) International Clinical Research Center, St. Anne's University Hospital, Brno (Czech Republic)
(c) Université Grenoble Alpes, CNRS, CEA, IRIG, LCBM, Grenoble (France)
(d) ESRF
References
[1] B. Lafumat et al., J. Appl. Crystallogr. 49, 1478-1487 (2016).
[2] V. Liskova et al., ChemCatChem 7, 648-659 (2015).
[3] D. Bednar et al., PLoS Comput. Biol. 11, e1004556 (2015).
[4] M. Musil et al., Nucleic Acids Res. 45, W393-W399 (2017).
[5] E. Chovancova et al., PLoS Comput. Biol. 8, e1002708 (2012).
[6] J. Stourac et al., Nucleic Acids Res. 47, W414-W422 (2019).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.