- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2008
- Structural biology
- SIRP structures explain paired-receptor specificity
SIRP structures explain paired-receptor specificity
The complex immune system of vertebrates must be tightly controlled in order to respond to pathogenic challenge without inducing unnecessary side effects. While soluble markers such as cytokines enable immune cells to localise to the site of infection, specific protein:protein interactions formed between cells fine-tune the immunological response.
The signal-regulatory proteins (SIRPs) are a family of cell-surface proteins expressed primarily on myeloid cells comprising three members: SIRP![]() , SIRPß and SIRP
, SIRPß and SIRP![]() [1]. While the extracellular regions of all three are very similar, the intracellular regions of these proteins are not alike and they convey very different signals to cells. SIRPα recognises CD47, a protein expressed on the surface of most cells that acts as a molecular ‘marker of self’, and formation of this CD47/SIRP
[1]. While the extracellular regions of all three are very similar, the intracellular regions of these proteins are not alike and they convey very different signals to cells. SIRPα recognises CD47, a protein expressed on the surface of most cells that acts as a molecular ‘marker of self’, and formation of this CD47/SIRP![]() interaction inhibits engulfment of cells by macrophages. SIRPß, on the other hand, does not recognise CD47 and interaction of SIRPß with its (unknown) extracellular ligand stimulates macrophage engulfment. While SIRP
interaction inhibits engulfment of cells by macrophages. SIRPß, on the other hand, does not recognise CD47 and interaction of SIRPß with its (unknown) extracellular ligand stimulates macrophage engulfment. While SIRP![]() recognises CD47 with lower affinity than SIRP
recognises CD47 with lower affinity than SIRP![]() , it is expressed on the surface of T-cells rather than macrophages and is thought to have no direct signalling function. The SIRPs thus typify the class of membrane protein families called “paired receptors”, which comprise several proteins with similar extracellular regions but radically different transmembrane/cytoplasmic regions with distinct (activating, inhibitory or neutral) signalling potentials.
, it is expressed on the surface of T-cells rather than macrophages and is thought to have no direct signalling function. The SIRPs thus typify the class of membrane protein families called “paired receptors”, which comprise several proteins with similar extracellular regions but radically different transmembrane/cytoplasmic regions with distinct (activating, inhibitory or neutral) signalling potentials.
The extracellular regions of SIRPs comprise three immunoglobulin (Ig) domains in a linear array. We had previously determined the structure of the first (amino-terminal) Ig domain of SIRP![]() [2]. Using site directed mutagenesis we showed that the interaction with CD47 was mediated by the N-terminal (apical) loops of SIRP
[2]. Using site directed mutagenesis we showed that the interaction with CD47 was mediated by the N-terminal (apical) loops of SIRP![]() , more closely resembling how antibodies recognise antigens rather than general Ig-domain mediated cell:cell contacts.
, more closely resembling how antibodies recognise antigens rather than general Ig-domain mediated cell:cell contacts.
To determine the precise mechanism by which SIRP![]() recognises its ligand, we solved the structure of the N-terminal SIRP
recognises its ligand, we solved the structure of the N-terminal SIRP![]() domain in complex with the sole extracellular Ig domain of CD47 in two crystal forms by molecular replacement using data collected at BM14 and ID23-2. Figure 74 shows the CD47/SIRP
domain in complex with the sole extracellular Ig domain of CD47 in two crystal forms by molecular replacement using data collected at BM14 and ID23-2. Figure 74 shows the CD47/SIRP![]() interface, which is unlike those seen for other moderate-affinity Ig domain mediated cell-surface interactions as it is highly convoluted, well fitting, and extensive, with a mutual involvement of loops from the amino-terminal ends of both SIRP
interface, which is unlike those seen for other moderate-affinity Ig domain mediated cell-surface interactions as it is highly convoluted, well fitting, and extensive, with a mutual involvement of loops from the amino-terminal ends of both SIRP![]() and CD47.
and CD47.
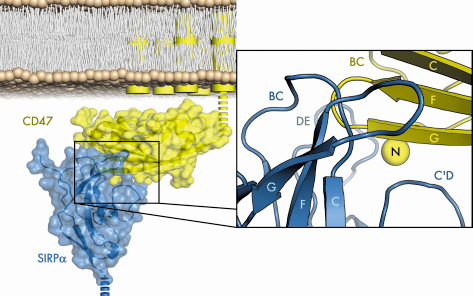 |
|
Fig. 74: The structure of human SIRP |
In contrast to SIRP![]() , SIRPß and SIRP
, SIRPß and SIRP![]() bind CD47 weakly or not at all, despite sharing > 90% sequence identity with SIRP
bind CD47 weakly or not at all, despite sharing > 90% sequence identity with SIRP![]() . We solved structures of the N-terminal domains of SIRP
. We solved structures of the N-terminal domains of SIRP![]() and two isoforms of SIRPß by molecular replacement using data collected at BM14. These structures demonstrate the exquisite selectivity of the SIRP interaction interface. Overall, the structures of SIRPs
and two isoforms of SIRPß by molecular replacement using data collected at BM14. These structures demonstrate the exquisite selectivity of the SIRP interaction interface. Overall, the structures of SIRPs ![]() , ß and
, ß and ![]() are very similar (Figure 75). However, for both isoforms of SIRPß specific amino acid differences with respect to SIRP
are very similar (Figure 75). However, for both isoforms of SIRPß specific amino acid differences with respect to SIRP![]() could be identified which result in the inability of SIRPß to bind CD47. Further, by introducing a single amino acid mutation in the sequence of one isoform of SIRPβ we were able to confer the ability to bind CD47 with an affinity approaching that of SIRP
could be identified which result in the inability of SIRPß to bind CD47. Further, by introducing a single amino acid mutation in the sequence of one isoform of SIRPβ we were able to confer the ability to bind CD47 with an affinity approaching that of SIRP![]() .
.
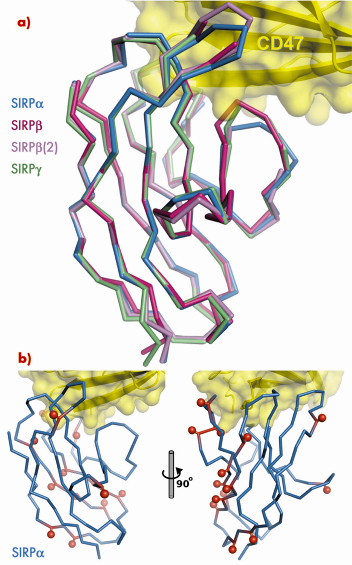 |
|
Fig. 75: a) Superposition of SIRPß and SIRP |
The N-terminal ligand-binding domain of SIRP![]() has much higher sequence variability than most other cell-surface receptors. Mapping this sequence variability onto the structure of the CD47/SIRP
has much higher sequence variability than most other cell-surface receptors. Mapping this sequence variability onto the structure of the CD47/SIRP![]() complex shows that most SIRPβa polymorphisms reside away from the interaction interface (Figure 75), implying that the sequence variability does not modulate the strength of the CD47 interaction. We propose that the driving force for this sequence variability might be to avoid recognition of SIRP
complex shows that most SIRPβa polymorphisms reside away from the interaction interface (Figure 75), implying that the sequence variability does not modulate the strength of the CD47 interaction. We propose that the driving force for this sequence variability might be to avoid recognition of SIRP![]() by pathogens, which would exploit the ability of SIRP
by pathogens, which would exploit the ability of SIRP![]() to down-regulate macrophage activity. An extension of the idea that SIRP
to down-regulate macrophage activity. An extension of the idea that SIRP![]() might be a target for pathogens is that SIRPß may have evolved to mimic the extracellular regions of SIRP
might be a target for pathogens is that SIRPß may have evolved to mimic the extracellular regions of SIRP![]() , its similar structure but opposite (activating) signalling activity serving to frustrate attempts by pathogens to exploit the inhibitory potential of SIRP
, its similar structure but opposite (activating) signalling activity serving to frustrate attempts by pathogens to exploit the inhibitory potential of SIRP![]() .
.
The structure of SIRP![]() in complex with CD47, together with the structures of SIRPß and SIRP
in complex with CD47, together with the structures of SIRPß and SIRP![]() in isolation, has shown us in molecular detail how this paired receptor binds its ligand and how subtle amino acid variations in the ligand-binding regions account for the exquisite ligand selectivity of the SIRP family. The partitioning of SIRP
in isolation, has shown us in molecular detail how this paired receptor binds its ligand and how subtle amino acid variations in the ligand-binding regions account for the exquisite ligand selectivity of the SIRP family. The partitioning of SIRP![]() polymorphisms away from the CD47 interaction interface suggests that pathogenic challenge may drive SIRP
polymorphisms away from the CD47 interaction interface suggests that pathogenic challenge may drive SIRP![]() polymorphism. Pathogen pressure on the inhibitory receptor provides a plausible raison d’être for the existence of paired receptors, with their similar extracellular structures but opposing signalling potentials.
polymorphism. Pathogen pressure on the inhibitory receptor provides a plausible raison d’être for the existence of paired receptors, with their similar extracellular structures but opposing signalling potentials.
Principal publication and authors
D. Hatherley, S.C. Graham, J. Turner, K. Harlos, D.I. Stuart, A.N. Barclay, Mol. Cell 31, 266 (2008).
University of Oxford (UK)
References
[1] A.N. Barclay, M.H. Brown, Nat. Rev. Immunol. 6, 457 (2006).
[2] D. Hatherley, K. Harlos, D.C. Dunlop, D.I. Stuart, A.N. Barclay, J. Biol. Chem. 282, 14567 (2007).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.