- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2016
- Structural biology
- Mode of action of FEN1 inhibitors revealed by X-ray crystallography and enzyme kinetic studies
Mode of action of FEN1 inhibitors revealed by X-ray crystallography and enzyme kinetic studies
The first inhibitor-bound structure of potential oncology target Flap endonuclease-1 (FEN1), together with biophysical studies, shed light on inhibitor mode of action: the cellularly active N-hydroxyurea compounds were found to block substrate entry to the catalytic site.
FEN1 belongs to the 5’-nuclease superfamily [1,2] and its main role is the removal of 5’ flaps generated during lagging-strand synthesis in DNA replication, thus it is essential for Okazaki fragment maturation [3,4]. FEN1 is overexpressed in multiple cancer types and has synthetic-lethal interactions with several genes frequently mutated in cancers. Due to its role in long-patch base excision repair, FEN1 is also a potential chemosensitising target [5,6,7].
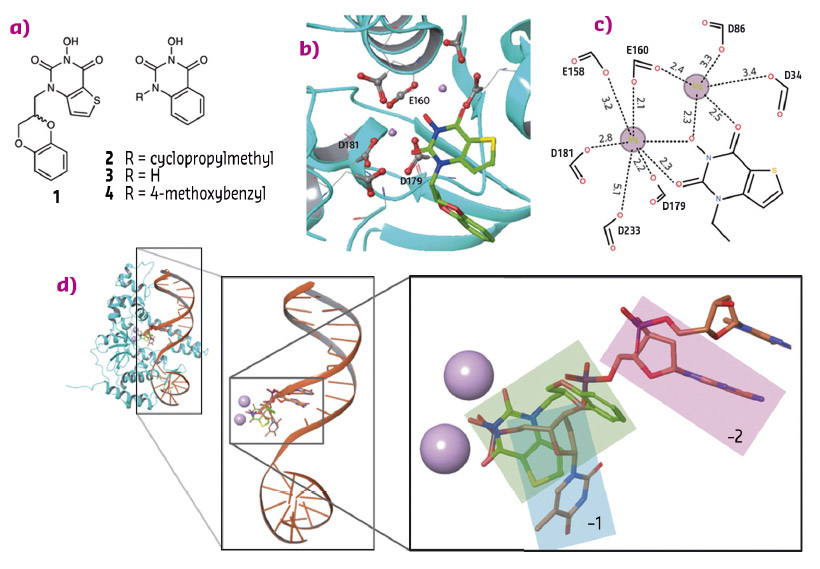
To establish the binding mode and possibly the mode of action of N-hydroxyurea inhibitors, human FEN1 was cocrystallised to form a ternary complex with Mg2+ and compound 1 (Figure 90a). The structure was solved following data collection at beamline ID29. Two Mg2+ ions, approx 4.5 Å apart, are coordinated by a number of acidic side chains that reside in the active site of the protein that forms an indentation of the kidney-bean shaped molecule (Figure 90b and 90c). The inhibitor molecule completes the coordination of the Mg2+ ions through its N-hydroxyurea moiety.
 |
|
Fig. 90: a) Compounds 1-4. b) Compound 1 bound to human FEN1 nuclease active site (PDB 5FV7). c) Schematic representation of the metal-coordination spheres. d) Structure of FEN1 in complex with product DNA (PDB 3Q8K) superimposed with FEN1 in complex with 1 (protein not shown). Metals are shown as pink spheres, the terminal 5' nucleotide (−1) is highlighted in the cyan box, the penultimate nucleotide of the product DNA (−2) is highlighted in pink, and 1 is highlighted in green. |
The ligand-bound structure suggests a possible mode of action. Superposition of FEN1 in complex with product DNA and this novel liganded structure shows the ligand occupying the location of the phosphate monoester of the DNA (Figure 90d). Biophysical studies were applied to characterise this type of inhibitor further.
Isothermal titration calorimetry (ITC) studies showed that Mg2+ was necessary for inhibitor binding, and replacing these with Ca2+ ions resulted in loss of ligand binding.
Kinetic experiments were undertaken to characterise FEN1 inhibition by 1, 2 and 4 vs DNA binding. All three compounds were able to bind both the DNA-free form and the substrate DNA-bound form of the enzyme. However, only compound 1 showed binding affinity to the DNA-complex comparable to the DNA-free state.
A fluorescence resonance energy transfer (FRET) assay was used to gain insight into the conformation of the DNA upon binding the inhibitor-bound form of FEN1. Both DNA binding sites required for catalytic activity were found to be occupied with and without the inhibitor. Using a tandem 2-aminopurine exciton pair preceding the cleavage site of the 5’-flap strand, CD spectra confirmed that the inhibitors prevented the base unpairing required for hydrolysis.
Compounds 1 and 4 could also be shown to be cellularly active in a cellular thermal shift assay (CETSA) in SW620 colon cancer cells in spite their different inhibition modes, with similar drug activity (EC50 values). However, there was a substantial drop-off compared to biochemical assays, probably due to high local concentrations of FEN1 in the nucleus during S-phase.
Principal publication and authors
Cellularly active N-hydroxyurea FEN1 inhibitors block substrate entry to the active site, J.C. Exell (a), M.J. Thompson (a), L.D. Finger (a), S.J. Shaw (a), J. Debreczeni (b), T.A. Ward (c), C. McWhirter (b), C.L. Siöberg (d), D. Martinez Molina (d), W.M. Abbott (b), C.D. Jones (e), J.W. Nissink (f), S.T. Durant (g) and J.A. Grasby (a), Nat Chem Biol. 12 815-821 (2016); doi: 10.1038/nchembio.2148.
(a) Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield (UK)
(b) Discovery Sciences, Innovative Medicines, AstraZeneca, Cambridge (UK)
(c) Bioscience, Oncology Innovative Medicines, AstraZeneca, Alderley Park, Cheshire (UK)
(d) Pelago Bioscience AB, Solna (Sweden)
(e) Chemistry, Oncology Innovative Medicines, AstraZeneca, Alderley Park, Cheshire (UK)
(f) Chemistry, Oncology Innovative Medicines, AstraZeneca, Cambridge (UK)
(g) Bioscience, Oncology Innovative Medicines, AstraZeneca, Cambridge (UK)
References
[1] J.A. Grasby et al., Trends Biochem. Sci. 37, 74–84 (2012).
[2] S.E. Tsutakawa et al., Cell 145, 198–211 (2011).
[3] S. Waga et al., J. Biol. Chem. 269, 10923–10934 (1994).
[4] R.A. Bambara et al., J. Biol. Chem. 272, 4647–4650 (1997).
[5] D.M. van Pel et al., PLoS Genet. 9, e1003254 (2013).
[6] J.L. Illuzzi et al., Curr. Med. Chem. 19, 3922–3936 (2012).
[7] J.-C. Hwang et al., PLoS One 10, e0139435 (2015).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.