- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2016
- Structural biology
- Structures of quinolinate synthase in complex with a substrate analogue, a key intermediate and product
Structures of quinolinate synthase in complex with a substrate analogue, a key intermediate and product
The enzyme NadA catalyses the synthesis of quinolinic acid (QA), the precursor of the universal nicotinamide adenosine dinucleotide cofactor. We report the crystal structures of NadA variant complexes with a substrate analog, the first reaction intermediate and the QA product, the latter two directly obtained from substrates.
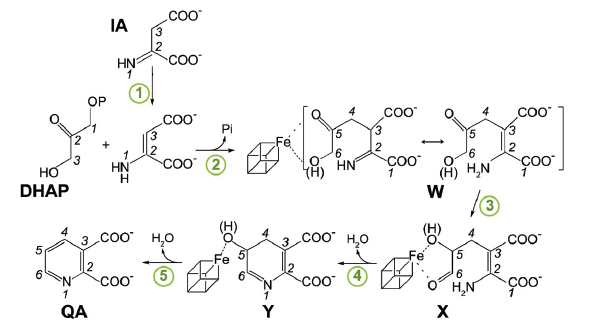
The adenine dinucleotide (NAD) cofactor is involved in many fundamental biochemical reactions through hydride transfer. Most prokaryotes synthesise NAD using the de novo II pathway that initially involves the condensation of iminoaspartate (IA) with dihydroxyacetone phosphate (DHAP) to generate quinolinic acid (QA) [1]. This reaction is catalysed by the [4Fe-4S]-containing enzyme quinolinate synthase NadA. Besides possibly transforming IA into its enamine tautomer (1 in Figure 86), NadA catalyses the condensation of this tautomer with DHAP (2), the aldo-keto isomerisation of the condensed intermediate (3) and two dehydration steps (4 and 5).
 |
|
Fig. 86: Proposed pathway of quinolinic acid synthesis by NadA. |
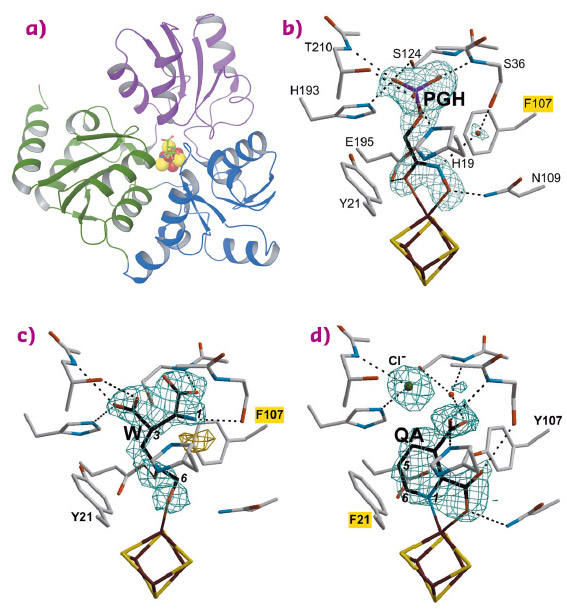
In 2014, we determined the holo crystal structure of Thermotoga maritima (Tm) NadA (Figure 87a) and described a long tunnel connecting its active site [4Fe-4S] cluster with the molecular surface [2]. Now, using X-ray data collected at beamlines BM30A, ID30B, ID23-1 and ID30A-3 (Massif-3), we have solved the structures of complexes of the TmNadA Y107F active site variant, which lacks QA synthetic activity, with (i) the DHAP analog phosphoglycolohydroxamate (PGH) at 1.45 Å resolution (Figure 87b) and (ii) the first condensation intermediate W at 1.9 Å resolution (Figure 86 and Figure 87c). The TmNadA Y107F-W complex (Figure 87c) was obtained by previously incubating the enzyme overnight anaerobically in a glove box with DHAP and oxaloacetic acid + ammonium ions (to generate IA non-enzymatically [3].
 |
|
Fig. 87: a) Ribbon depiction of the quinolinate synthase NadA with bound W and colour-coded domains and omit electron density maps and atomic models corresponding to b) PGH; c) W and d) QA complexes. Mutated residues are highlighted in yellow. |
Our refined model of W has the carbonyl oxygen at C5 and a C6 terminal alkoxide group implying that the aldo-keto isomerisation does not occur in TmNadA Y107F thus explaining its inability to synthesise QA. Indeed, cyclisation to yield Y from X requires a C6 terminal aldehyde [4,5] (Figure 86). W displays several of the features expected for a condensation product/reaction intermediate. First, its two carboxylic groups bind TmNadA as the IA analog malate does in the P. horikoshi NadA structure [6]. Second, both the substrate analog PGH and W bind to the [4Fe-4S] cluster with their terminal –O(H) moieties and their C=O groups. Third, the structure of W also shows that, as previously suggested, the condensation of IA with DHAP involves the elimination of the phosphate group of the latter (Figure 86 and Figure 87c). Fourth, W has its N atom well oriented for its nucleophilic attack on C2 and subsequent cyclisation (Figure 86 and Figure 87c) [6].
Using the slowly-reacting TmNadA Y21F variant, we have reacted the same substrates to form a crystalline complex between this protein and the QA product and solved it at 1.9 Å resolution (Figure 87d).
In summary, our study shows that (i) the condensation of IA with DHAP is coupled to dephosphorylation; (ii) Tyr107 is most likely involved in the aldo-keto isomerisation of W to X and in imino-enamine tautomerisation; (iii) after the second dehydration, helped by Tyr21, the QA product rotates inside the NadA active site forming a bidentate complex with the unique Fe ion of the [4Fe-4S] cluster. Which mechanism is responsible for QA release from NadA will be the subject of future investigations.
Besides the fundamental interest of elucidating the catalytic mechanism of NadA, our structural studies constitute a solid basis for the design of novel antibiotics targeted against two human pathogens that use exclusively the de novo II pathway of NAD synthesis: Helicobacter pylori, which causes gastric ulcers and cancer and Mycobacterium leprae, the causal agent of leprosy.
Principal publication and authors
Crystal structures of quinolinate synthase in complex with a substrate analogue, the condensation intermediate, and substrate-derived product, A. Volbeda (a), C. Darnault (a), O. Renoux (b), D. Reichmann (b), P. Amara (a), S. Ollagnier-de-Choudens (b) and J.-C. Fontecilla-Camps (a), J Am Chem Soc 138, 11802 (2016); doi: 10.1021/jacs.6b05884.
(a) Metalloproteins Unit, Institut de Biologie Structurale, Grenoble (France)
(b) BIG/CBM/Biocat, CEA-Grenoble (France)
References
[1] S.Y. Gerdes et al., J. Bacteriol. 184, 4555 (2002).
[2] M.V. Cherrier et al., J. Am. Chem. Soc. 136, 5253 (2014).
[3] S. Nasu et al., Biophys. Res. Commun. 101, 533 (1981).
[4] D. Reichmann et al., Biochemistry 54, 6443 (2015).
[5] N. Suzuki et al., Biochim. Biophys. Acta 304, 309 (1973).
[6] S. Nasu et al., J. Biol. Chem. 257, 626 (1982).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.