- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2014
- Dynamics and extreme conditions
- A new crystal phase of ferrocene
A new crystal phase of ferrocene
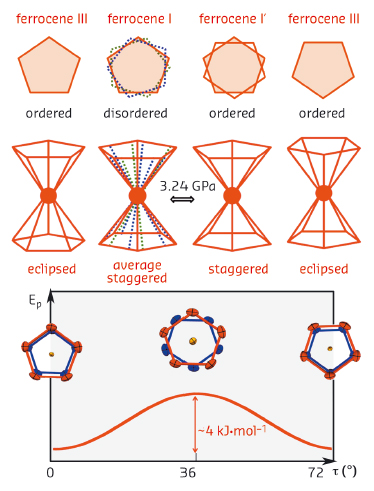
In 1951 the serendipitous discovery of ferrocene, FeC10H10, bis(cyclopentadienyl)iron(II), the first and one of the most stable metallocenes, ignited the ‘explosion’ of organometallic chemistry [1,2]. Since then, thousands metallocenes and their derivatives have been synthesised and used as catalysts in chemical laboratories and in technological processes [3,4]. The proposed sandwich-like structure of the ferrocene molecule, consisting of two parallel cyclopentadienyl (Cp) rings with Fe2+ ion sandwiched in-between, was soon confirmed by spectroscopic and X-ray diffraction studies. The structure of ferrocene has been determined independently in several laboratories, but its molecular conformation and interactions remained unclear. Under normal conditions, ferrocene polymorph I is monoclinic with staggered cyclopentadienyl rings, i.e. their torsion angles τ equal to 36º + n·72º (τ is defined as torsion C-cCp···cCp’-C’, where unprimed and primed symbols refer to two Cp-rings of one molecule, cCp and cCp’ are the rings centroids, and n is an integer). The staggered conformation is energetically unfavoured and high-resolution neutron-diffraction measurements revealed structural models with Cp-rings disordered in two or three sites (Figure 83).
 |
|
Fig. 83: Schematic illustration of the conformers of ferrocene molecule and their potential energy function (Ep). The torsion angle τ describes the mutual orientation of the cyclopentadienyl rings (Cp), as defined in the text. The dotted green and blue lines in the drawing of the molecule in phase I indicate the positions of disordered Cp-rings. |
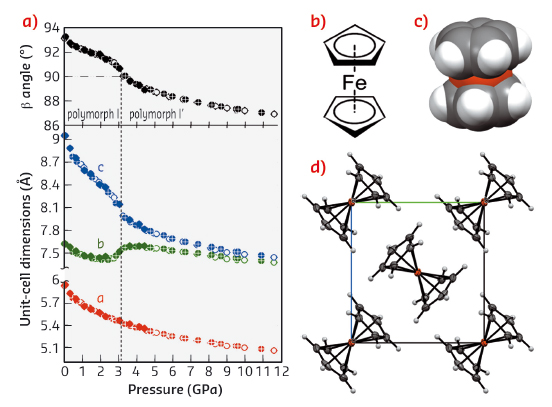
Below 164 K, ferrocene I transforms into the triclinic form II, where the Cp-rings of two independent molecules are each distorted by about 9º off the energetically favoured eclipsed conformation (τ = 0º, Figure 83). Dunitz et al. [5] crystallised ferrocene below 98 K and obtained its orthorhombic polymorph III with the Cp-rings eclipsed at τ equal to 0º. These results suggested that the unfavoured staggered conformation in ferrocene I is promoted by intermolecular interactions. However, under normal conditions, these crystal cohesion forces are too weak to completely stabilise the molecular rings at the unfavourable staggered conformation. The intermolecular interactions of ferrocene I have been enhanced by high pressure in our single-crystal X-ray diffraction experiment performed at beamline ID09A. We found that ferrocene I undergoes an isostructural phase transition at 3.24 GPa (Figure 84), preserving the crystal unit cell and symmetry of space group P21/n.
 |
|
Fig. 84: a) Unit-cell dimensions of ferrocene phases I and I’ compressed up to 11.6 GPa. The vertical dashed line marks the transition between phases I and I’ at 3.24 GPa. The filled circles represent laboratory data, crossed circles and empty circles indicate the compression and decompression runs measured at beamline ID09A, respectively; b) structural formula of the molecule in the staggered conformation, used in most chemistry textbooks; c) space-filling model of the ferrocene molecule in phase I; and d) molecules of ferrocene located at the inversion centres in the monoclinic polymorph I’. |
Therefore, this new high-pressure phase of ferrocene has been denoted as phase I’. In its structure, the molecules are ‘pushed’ by intermolecular forces into the staggered conformation and the disorder of Cp-rings is eliminated. This result is consistent with the occurrence of the average staggered conformers unfavoured by 4 kJmol-1 in ferrocene I. In contrast, this pressure-induced phase transition is in many respects different to most other order-disorder phase transitions. Usually in high-temperature phases molecules or ions are disordered, in two or more equivalent or nearly equivalent sites. On lowering temperature, the disorder in the structure is eliminated when Cp-rings reside in only one of the sites. Thus, below 164 K the disordered ferrocene I transforms into ferrocene II with two independant molecules ordered. We were able to show that high pressure has a very different effect on the crystal structure, and that in ferrocene I’ it does not favour any of the disordered sites, but the average and the least energetically-favoured conformation. We were also able to explain the U-turn shaped compression of the unit-cell parameter b, by the compensation of the shortest intermolecular distances in the compressed structure. However, several intriguing observations could be made about the measured data. The discontinuities in unit-cell parameters occur when angle β, obtuse in ferrocene I, passes through 90º and it becomes acute in phase I’. The β = 90º coincides with the point of structural transformations when the first and second shortest intermolecular contacts between the centres of molecules in ferrocene I become equal and then reversed in phase I’. In these respects, the structure of ferrocene phases I and I’ resembles a mechanical box with meshing gears regulated by their positions and distances. Our study shows that a lot of information is still missing for a complete understanding of the basic properties of prototypical compounds at their molecular level, and that high pressure studies provide a new perspective for their investigation.
Principal publication and authors
D. Paliwoda (a,b), K. Kowalska (a), M. Hanfland (b) and A. Katrusiak (a), J. Phys. Chem. Lett. 4, 4032–4037 (2013).
(a) Faculty of Chemistry, Adam Mickiewicz University, Poznan (Poland)
(b) ESRF
References
[1] T.J. Kealy and P.L. Pauson, Nature 168, 1039−1040 (1951).
[2] P.L. Pauson, J. Organomet. Chem. 637−639, 3−6 (2001).
[3] H. Werner, Angew. Chem. Int. Ed. 51, 6052−6058 (2012).
[4] G.B. Kauffman, J. Chem. Educ. 60, 185−186 (1983).
[5] P. Seiler and J.D. Dunitz, Acta Crystallogr. Sect B 38, 1741−1745 (1982).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.