- Home
- News
- Spotlight on Science
- Shedding synchrotron...
Shedding synchrotron light on noble metal nanocatalyst strain dynamics
12-10-2021
X-ray diffraction at beamline ID31 has revealed the interactions between nanocatalysts and electrochemical species during operation in liquid electrolytes. The strain in nanocatalysts is shown to describe both their absorption and adsorption trends, which are critical for understanding their performance and stability.
The eventual future of a hydrogen economy relies strongly on the development of key electrochemical conversion devices, such as proton exchange membrane water electrolysers (PEMWE, for the production of hydrogen from electricity) and proton exchange membrane fuel cells (PEMFC, for the production of electricity from hydrogen). In both cases, catalyst materials play a major role in device cost, performance and lifetime. Consequently, there is an urgent need to design more effective catalysts.
In the present case of heterogeneous catalysis (where the reactions occur on the catalyst surface), the most promising approach in materials design follows the Sabatier principle: the ability of the surface to bind adsorbates, and the strength of the bonds, define the reaction thermodynamics and kinetics. In this respect, the catalyst surface chemistry and structure (crystallographic orientation of the facets and/or strain) are used as levers to optimise performance.
However, it is largely accepted that the structure of PEMWE anode and PEMFC cathode catalysts is altered during long-term operation due to harsh (electro)chemical conditions. Pioneer work conducted at beamline ID31 has previously revealed that platinum (Pt) nanoparticle strain is also generally a function of the electrode potential [1]. This was qualitatively rationalised as a consequence of the adsorption of various molecules on the Pt surface, which opened fundamental questions: do the observed structural modifications of the catalyst provide critical information about catalyst adsorption trends, and do they impact the properties of the catalyst?
In recent collaborative work, researchers from ID31, the University of Grenoble-Alpes and the Technical University of Berlin investigated strain dynamics for Pt and palladium (Pd) nanocatalysts in liquid electrolytes. The work demonstrates the power of the new ESRF-EBS source to reveal the microstructural information of device-relevant nanomaterials in a liquid electrochemical environment under operando conditions with unprecedented quality and temporal resolution (Figure 1a).
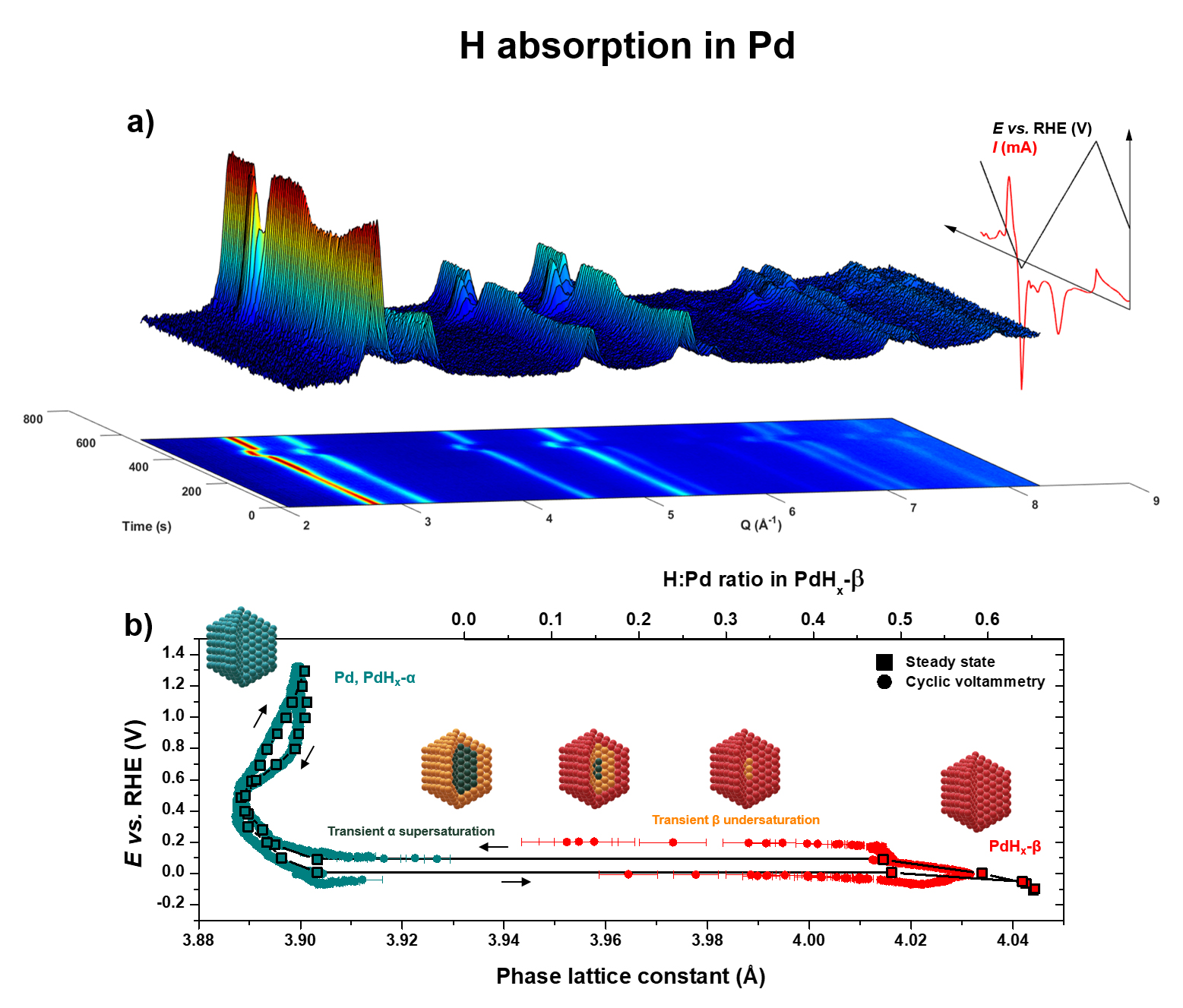
Fig. 1: Monitoring hydrogen absorption in Pd. a) 3D representation and corresponding 2D projection of wide-angle X-ray scattering (WAXS) pattern intensities (1 over 20) plotted as a function of time and the momentum transfer Q recorded during a Pd cyclic voltammetry experiment in a N2-purged 0.1 M NaOH electrolyte at room temperature. The evolution of the electrochemical potential applied, and electric current measured, are also represented on the same time scale. b) PdHx α ↔ β phase transition lattice constant hysteresis at the steady state (square symbols interpolated by black lines) measured after 60 s hold at various potentials, superimposed with the data from cyclic voltammetry at 5 mV s-1 (circle symbols). The temporal resolution is 333 ms.
Beyond the technical showcase, the study confirms that the bulk microstructures (lattice constant, crystal phase or coherence) of such device-relevant catalysts with enhanced surface-to-volume ratio are permanently modified by the electrochemical environment of their application systems (Figure 1a and Figure 2a for Pd and Pt, respectively). The ability of Pd to absorb hydrogen and to form Pd hydrides is already largely documented. However, monitoring the structure of Pd nanoparticles during cyclic voltammetry and potential staircase allowed the researchers to unveil the electrochemically driven Pd hydride phase transition, which was, until now, mostly investigated in the gas phase. The fine X-ray diffraction patterns measured under operando conditions provided the first detection of theoretically predicted supersaturated and undersaturated metastable states, involved in a core-shell mechanism of the phase transition (Figure 1b).
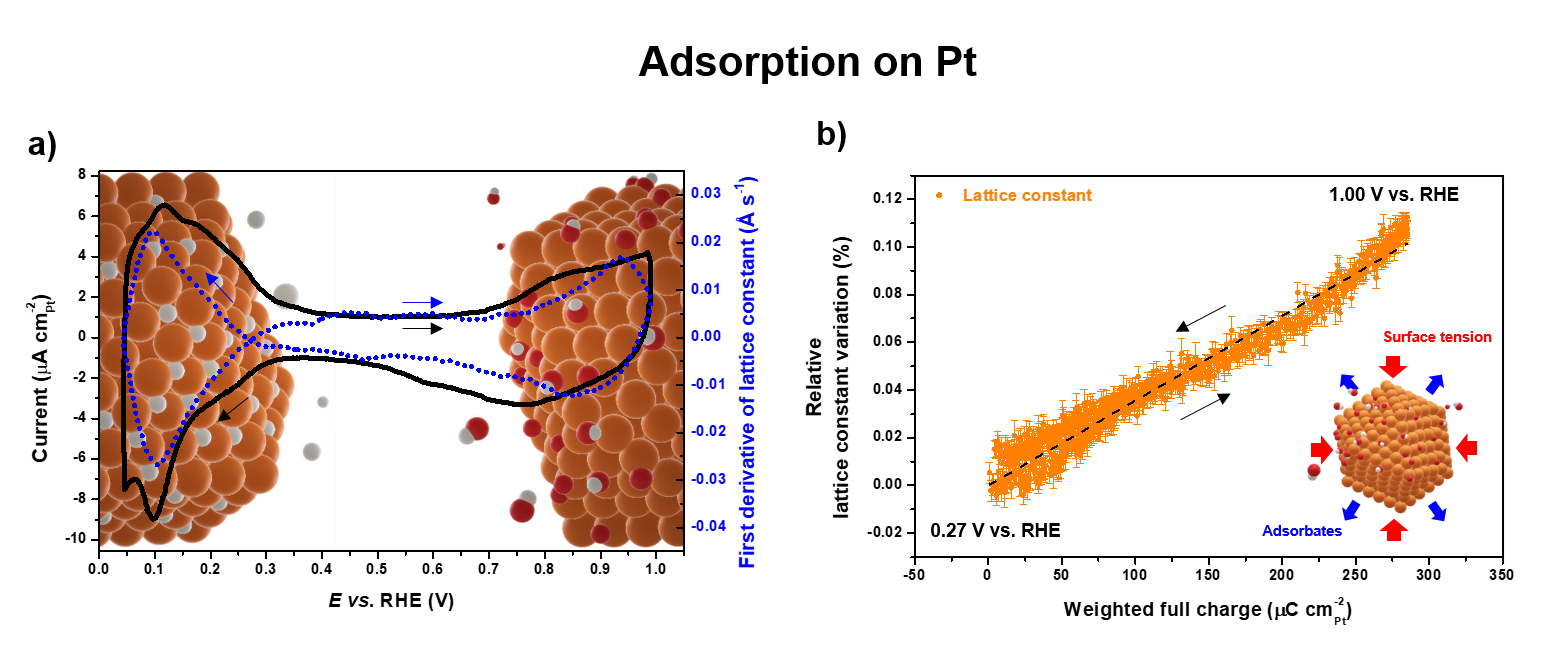
Fig. 2: Monitoring adsorption on Pt. a) Cyclic voltammograms (solid line) and first derivative of the lattice constant (dashed line) of Pt/C in N2-purged 0.1 M HClO4 recorded at room temperature with a potential sweep rate of 5 mV s-1. b) Linear regression (dashed line) between lattice constant variation and adsorbate-related electrical charge.
In addition, monitoring the structure of Pt/C during cyclic voltammetry allowed the establishment of a near-linear correlation between the expansion of the bulk lattice parameter of Pt nanoparticles and their oxide surface coverage (Figure 2b). Since the latter is known to be a descriptor of catalyst activity toward numerous reactions (oxygen reduction and fuel oxidation) and stability (oxidation and dissolution), the measurement of the lattice parameter according to this simple diffraction approach allows experimental access to this descriptor during measurements under operando conditions in device-relevant sample environments. This constitutes a major advantage compared to spectroscopic techniques, and is expected to find a wide range of applications.
Finally, in light of previous Density Functional Theory (DFT) calculations, this work shows that the strain variation amplitude associated with molecular adsorption is not expected to significantly alter the electro-catalytic properties of nanoparticles in the case of the Oxygen Reduction Reaction (ORR) on Pt.
Principal publication and authors
Electrochemical Strain Dynamics in Noble Metal Nanocatalysts, R. Chattot (a), I. Martens (a), M. Mirolo (a), M. Ronovsky (a), F. Russello (a), H. Isern (a), G. Braesch (b), E. Hornberger (c), P. Strasser (c), E. Sibert (b), M. Chatenet (b), V. Honkimäki (a), J. Drnec (a), J. Am. Chem. Soc. (2021); https://doi.org/10.1021/jacs.1c06780
(a) ESRF
(b) Univ. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, Grenoble INP, LEPMI, Grenoble (France)
(c) Electrochemical Energy, Catalysis and Material Science Laboratory, Department of Chemistry, Technische Universität Berlin, Berlin (Germany)
References
[1] I. Martens et al., ACS Appl. Energy Mater. 2 (11), 7772-7780 (2019).
Top image: Credit: R. Chattot et al.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.