- Home
- News
- Spotlight on Science
- X-ray crystallography...
X-ray crystallography reveals the structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ)
03-02-2022
In response to cellular stress, the tumour suppressor p53 activates genes critical for cancer prevention. In more than 50% of human cancers, mutations in p53 lead to its inactivation. A small molecule, methylene quinuclidinone (MQ), reactivates mutant p53. Crystal structures of p53 mutants bound to MQ, obtained by X-ray crystallography at beamlines ID23, ID29 and ID30A-3, reveal the reactivation mechanism of cancer-related p53.
The p53 protein is a tumour suppressor that, in response to cellular stress, acts as a transcription factor by binding as a tetramer (a dimer of dimers) to a wide range of DNA response elements, activating various events including DNA repair, cell-cycle arrest, senescence or apoptosis, all being critical to cancer prevention.
The human p53 protein is 393 residues long and contains several major functional domains, of which the N-terminus contains a transactivation domain, the core domain contains a sequence-specific DNA binding domain (p53DBD) and the C-terminus incorporates tetramerisation and regulatory domains. Of these domains, only the DNA-binding and the oligomerisation domains are structured, whereas other parts of the protein are intrinsically disordered.
In more than half of human cancers, mutations are observed in the TP53 gene, leading to loss of wild-type p53 function. Over 90% of the tumorigenic mutations are found in the DNA-binding core domain, spanning nearly 200 residues (94–293). Among these mutations, six are referred as hotspots due to their high frequency (nearly 30%) in various types of cancer. Mutations at positions R248 and R273, referred as DNA-contact mutations, lead to loss of direct p53-DNA interactions. Mutations at positions R175, G245, R249 and R282, referred as structural mutations, lower p53 stability and modify its folding state in a mutation-specific manner from local distortions to global denaturation.
Because of its key role in cancer development, mutant p53 has been a target for restoring wild-type function to mutant p53 by small drug molecules. Among the most clinically advanced, eprenetapopt (APR-246/PRIMA-1MET) is a small molecule that, following conversion into its biologically active compound, methylene quinuclidinone (MQ), reactivates mutant p53 by binding covalently to cysteine residues in p53. Two enantiomers may be formed upon MQ binding to a cysteine thiol group. This reaction is highly reversible; hence, the selection of either enantiomer at a specific cysteine residue varies, depending on stabilising interactions with the molecular surrounding.
Previous X-ray crystallography studies have uncovered the structural basis of DNA recognition by p53 core-domain tetramers [1-3], and the functional loss of p53 hotspot mutants and their reactivation by suppressor mutations [4-5]. In the present work, to uncover the structural basis of rescuing p53 mutants by MQ, X-ray crystallography was carried out at beamlines ID23-1, ID23-2, ID29 and ID30A-3, and an in-house facility, to obtain high-resolution structures of the core domains of mutant and wild-type p53 bound to MQ in their free state and/or bound to their DNA response elements.
The wild-type and mutant p53DBD, including two DNA-contact mutants, R273H and R273C, and a structural mutant, R282W, were studied. Both co-crystallisation and soaking approaches were tried in a systematic manner to obtain crystals of p53 and/or p53-DNA complexes bound to MQ. The concentration of MQ played a key role in the process: typically, too-low concentrations led to MQ-free structures, whereas too-high concentrations led to poorly diffracting crystals. Yet, a large excess of MQ was required to obtain crystals of p53DBD bound to MQ owing to the high reversibility of the covalent binding. Fourteen new crystal structures were determined, allowing for detailed analysis of the effects of MQ on mutant and wild-type p53 and their complexes with DNA.
Click image to enlarge
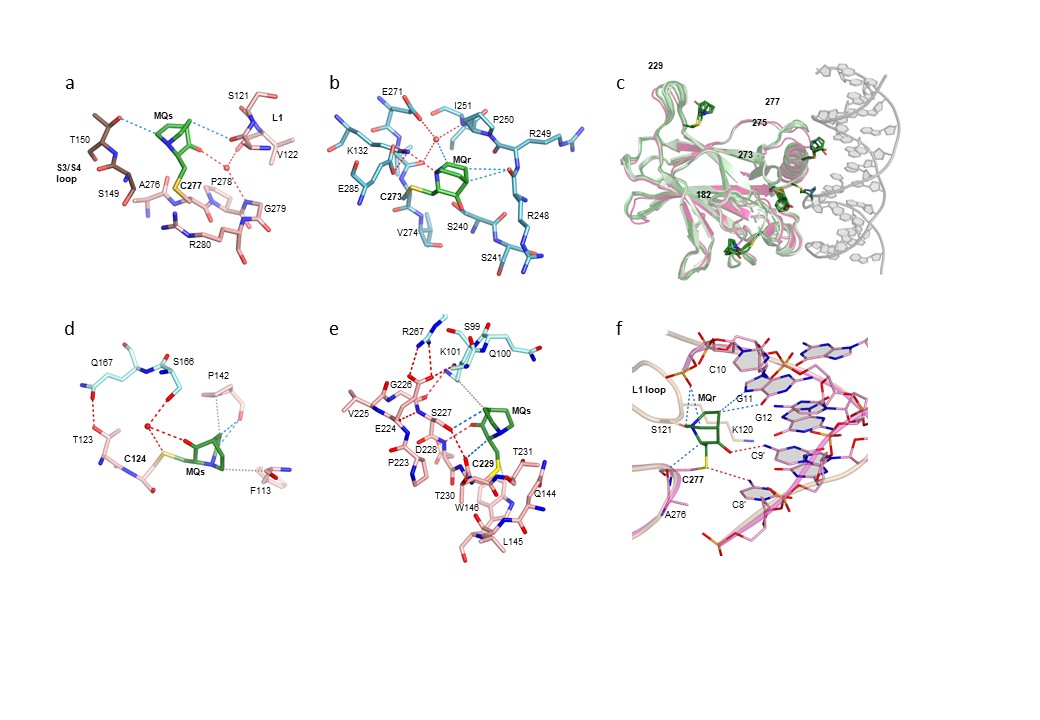
Fig. 1: MQ bound to cysteines in the DNA-free and DNA-bound structures of mutant p53DBD. a) MQ bound to C277 in one monomer of R273H-MQ structure. b) MQ bound to C273 in one monomer of R273C-MQ structure. c) Superposition of all monomers from R273C-MQ (I) and (II) structures (each incorporating four monomers in the asymmetric unit) shown in light green, onto a monomer from wt-p53DBD tetramer bound to DNA (based on PDB 5MCV) shown in magenta and grey. This view demonstrates that MQ bound to C182, C229, C273, and C277 are compatible with DNA binding, but not C275. d) MQ bound to C124 in R282W-DNA-MQ structure. e) MQ bound to C229 in one monomer of R282W-DNA-MQ structure. f) MQ bound to C277 in one monomer of R273H-DNA-MQ structure. Also shown: cartoon representations of the DNA backbones of the MQ-modified (magenta) and the MQ-free (light brown) R273H-DNA complexes. The corresponding enantiomers of MQ (shown in green) are assigned as MQs and MQr. Neighbouring monomer is shown in brown in (a), and blue in (d) and (e). The various MQ-Cys adducts contribute to local stabilisation of p53 regions around the modified sites and to DNA binding by hydrogen bonds, CH···O and VdW interactions (shown as dotted lines in red, blue and grey, respectively). In all p53DBD-DNA tetrameric complexes, the four monomers are related by symmetry.
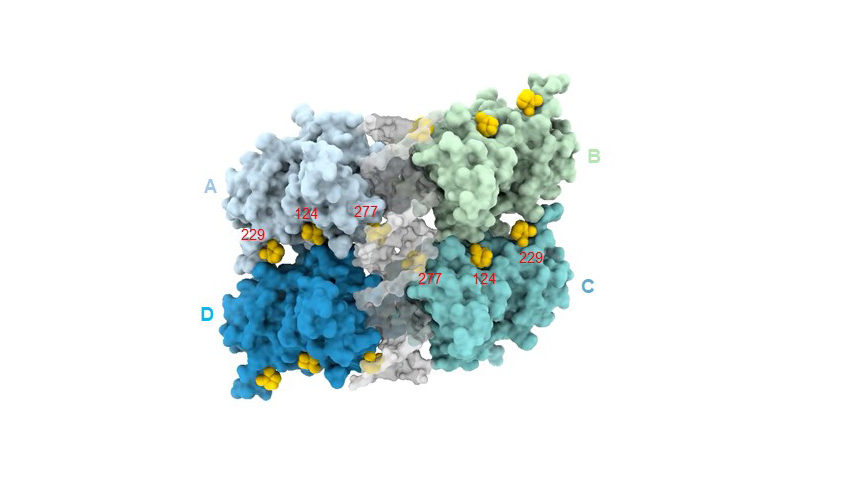
The detailed structural analysis shows that out of the 10 native cysteines of the core domain, only five residues (C124, C182, C229, C275, C277), as well as the hotspot mutation product C273, were found modified in the various DNA-free structures, all located at the surface of the protein (Figures 1a-c). MQ selectivity is higher in the DNA-bound state of p53; only three MQ-modified cysteines (C124, C229 and C277) were observed in the p53-DNA-MQ structures of both mutant and wild-type p53 (Figures 1d-f); MQ-C124 and MQ-C229 support the inter-dimer interface and MQ- C277 stabilises the protein-DNA interface (Figure 2). In both DNA-free and DNA-bound p53, MQ binding to cysteines appears variable and dynamic, showing a large diversity in its interaction modes.
Click image to start video
Fig. 2: Tetramer assembly of mutant p53DBD bound to DNA and MQ based on the structure of R282W-DNA-MQ (PDB ID 7B4E). p53 monomers are shown in blue, cyan, light blue and green, DNA in grey and MQ in yellow.
The present study reveals the role played by MQ in stabilising p53 and its interaction with DNA, thus providing a structural framework for the design of new drug molecules to target specific p53 cysteines for potential cancer therapy.
Principal publication and authors
Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ), O. Degtjarik (a,d), D. Golovenko (a,e), Y. Diskin-Posner (b), L. Abrahmsén (c), H. Rozenberg (a), Z. Shakked (a), Nat. Commun. 12(1), 7057 (2021); https://doi.org/10.1038/s41467-021-27142-6
(a) Department of Chemical and Structural Biology, Weizmann Institute of Science (Israel)
(b) Department of Chemical Research Support, Weizmann Institute of Science (Israel)
(c) Aprea Therapeutics AB (Sweden)
(d) Present address: Astbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology, University of Leeds (UK)
(e) Present address: University of Massachusetts Medical School, RNA Therapeutics Institute, Worcester, MA (USA)
References
[1] M. Kitayner et al., Mol. Cell 22(6), 741-753 (2006).
[2] M. Kitayner et al., Nat. Struct. Mol. Biol. 17, 423–429 (2010).
[3] D. Golovenko et al., Structure 26(9), 1237-1250 (2018).
[4] O. Suad et al., J. Mol. Biol. 385, 249–265 (2009).
[5] A. Eldar et al., Nucleic Acids Res. 41, 8748-8759 (2013).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.