- Home
- News
- Spotlight on Science
- The transition state...
The transition state of enzyme phosphoryl transfer revealed by magnesium triflouride
23-04-2010
The transfer of phosphoryl groups is probably the most important enzyme-catalysed reaction in biology. All organisms use phosphate in the form of ATP to store and transmit energy but phosphoryl transfer is also used to control processes as diverse as cell signalling, regulation of cellular division, membrane structure and enzyme function. Consequently, a huge number of enzymes, up to 10% of the human genome, have evolved to catalyse phosphoryl transfer. Dissecting how these enzymes catalyse the reaction is of vital importance in understanding the cellular processes they regulate and perhaps designing drugs to target specific pathways. Structural studies of two analogues of the transition state of phosphoryl transfer, magnesium trifluoride and aluminium tetrafluoride, using high resolution X-ray crystallography in combination with 19F-NMR, has provided exceptional details of the mechanism of the reaction in two archetypal phosphoryl transfer enzymes.
Share
The manipulation and stability of phosphate mono- and di-esters underpin all of biology as they are involved in the storage and transmission of genetic information, energy transfer, signalling and cellular differentiation. They are remarkably resistant to spontaneous hydrolysis (DNA has been sequenced from the mummy of Tutenkhamun after 3320 years) and yet enzyme-catalysed phosphoryl transfer is sufficiently fast to support a large array of biological processes [1]. While the general nature of this reaction has been established by sterochemical studies, analysis of nucleophile and leaving group dependencies and a large body of structural information, details of the transition state remain elusive. The reaction is thought to proceed via a trigonal, planar phosphoryl species, PO3-, but the nature of this moiety's interactions within a protein are poorly understood. Recently, magnesium trifluoride (MgF3-) has emerged as a surrogate for PO3- in enzyme-catalysed phosphoryl transfer transition states [2,3]. We combined solution NMR and high resolution X-ray crystallography to study the enzymes β-phosphoglucomutase (β-PGM) and human phosphoglycerate kinase (hPGK), demonstrating that MgF3- is a highly sensitive probe of the electrostatic and hydrogen bonding environment of the transition state of phosphoryl transfer and confirming experimentally the importance of charge balance in the catalysis of the reaction.
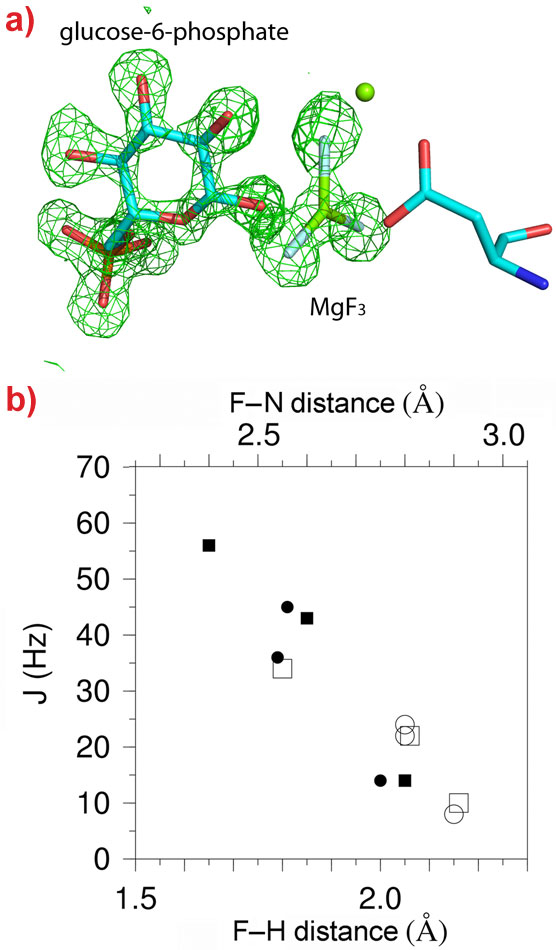
19F-NMR provides detailed information on the environment of fluoride nuclei but no information on their positioning within the active site of an enzyme. We combined the resonances obtained from β-PGM (an enzyme that transfers a phosphoryl group from carbon-1 to carbon-6 of glucose) in complex with glucose-6-phosphate and MgF3- with a high resolution (1.3 Å) X-ray crystal structure of the same complex (Figure 1a). This allowed an exact structural definition of the MgF3- species and comparison between atomic positions and 19F-NMR fluorine resonances to be made. Proton distributions in the vicinity of the fluorides can be determined from hydrogen/deuterium primary isotope shifts, 19F-1H Nuclear Overhauser Effects (NOEs) and scalar coupling associated with hydrogen bonding (Figure 1b). These measurements provide a picture of the charge distribution “seen” by the transition state, and the H-bonding interactions, that cannot be determined from the crystal structure alone.
 |
|
Figure 1. The structure of the β-PGM-MgF3-G6P complex. a) A difference Fourier map (green mesh contoured at 3σ) showing the positions of glucose-6-phosphate and MgF3- in the 1.3 Å crystal structure of the complex. b) Correlation plot showing the relationships between the spin coupling constants JHF (filled symbols) and JNF (open symbols) couplings with the corresponding internuclear distances derived from structures of the β-PGM-MgF3-G6P (circles) and β-PGM-AlF4-G6P (squares) complexes. |
This technique was also applied to human phosphoglycerate kinase (hPGK), the enzyme responsible for the first energy-generating step in glycolysis and an important target in the treatment of cancer and retroviral diseases such as AIDS and hepatitis. In complex with the substrates ADP, phosphoglycerate (3PG) and MgF3-, three distinct fluoride resonances are observed (Figure 2a). The crystal structure of this complex was solved to 1.47 Å resolution and revealed for the first time the active conformation of the protein with a catalytic triad of positively charged residues surrounding the transition state analogue (Figure 2b). The active state of the enzyme implied that neutralising the negative charge on the transition state was an important feature of catalysis. In order to test this theory, a similar complex, substituting MgF3- with aluminium tetrafluoride, AlF4-, was studied both in the natural enzyme and in a mutant enzyme where one of the important positive charges had been removed (K219). 19F-NMR of these systems revealed that one of the fluoride resonances is lost upon deletion of the positive charge (Figure 2a). The X-ray crystal structures of these complexes show that when the charged residue is removed, one of the fluorine atoms coordinating the aluminium is replaced with a water molecule, thus maintaining the balance of charge in the active site. Charge balance is likely to be important in transition state stabilisation by phosphoryl transfer enzymes because at neutral pH the attack of a dianionic phosphate group by a negatively charged nucleophile is extremely unfavorable. These results imply that enzymes have evolved to counteract the repulsion through tuning the active site to the charge of the transition state.
 |
|
Figure 2. a) 19F-NMR spectra of (A) the hPGK-3PG-MgF3-ADP TSA complex (FA= -153, FB = -156, F1 = -161 ppm). The most upfield F resonance is assigned as F1. The assignment of the other 19F resonances is ambiguous and therefore the resonances are labeled alphabetically from left to right on the basis of chemical shift. Free fluoride in solution resonates at -119 ppm. (B) the hPGK-3PG-AlF4-ADP complex (FA = -136, FB = -138, FC = -141, F1= -146 ppm). Again, the most upfield 19F resonance is assigned as F1. The other resonances are labeled alphabetically from left to right on the basis of chemical shift and do not necessarily indicate equivalence of nuclei between the spectra. (C) the hPGK(K219A)-3PG-AlF3-ADP complex (FA = -135, FB = -137, F1 = -143 ppm). b) Electron density in the active site of hPGK-3PG-MgF3-ADP complex. The difference electron density for MgF3 before its inclusion in the model is shown as a green mesh contoured at 3σ. The electron density for the 3PG and ADP ligands is shown as a blue mesh contoured at 1.5σ. |
References
[1] M.W. Bowler, et al., New J. Chem., 34, 784-794 (2010).
[2] D.L. Graham, et al., Chem. Biol., 9, 375–381 (2002).
[3] N.J. Baxter, et al., Proc. Natl. Acad. Sci. USA, 103, 14732-14737 (2006).
Principal publication and authors
N.J. Baxter (a), M.W. Bowler (b), T. Alizadeh (a), M.J. Cliff (a), A.M. Hounslow (a), B. Wu (c), D.B. Berkowitz (c), N.H. Williams (d), G.M. Blackburn (a), and J.P. Waltho (a,e), Atomic details of near-transition state conformers for enzyme phosphoryl transfer revealed by MgF3- rather than by phosphoranes, Proc. Natl. Acad. Sci., 107, 4555-4560 (2010);
M.J. Cliff (a), M.W. Bowler (b), A. Varga (f), J.P. Marston (a), J. Szabo (f), A.M. Hounslow (a), N.J. Baxter (a,e), G.M. Blackburn (a), M. Vas (f), and J.P. Waltho (a,e), Transition state analogue structures of human phosphoglycerate kinase reveal the dominance of charge balance in catalysis, J. Am. Chem. Soc., 132, 6507-6516 (2010).
(a) Department of Molecular Biology and Biotechnology, University of Sheffield (UK)
(b) ESRF
(c) Department of Chemistry, University of Nebraska-Lincoln (USA)
(d) Centre for Chemical Biology, Department of Chemistry, University of Sheffield (UK)
(e) Faculty of Life Sciences and Manchester Interdisciplinary Biocentre, University of Manchester (UK)
(f) Institute of Enzymology, Biological Research Center, Hungarian Academy of Sciences, Budapest (Hungary)
Top image: The transition state of phosphoryl transfer in human phosphoglycerate kinase revealed by magnesium triflouride (Image credit: M. Bowler)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.