- Home
- News
- Spotlight on Science
- The birth of micelles...
The birth of micelles revealed by synchrotron light
20-05-2009
Spontaneous micellisation of block copolymers in selective solvents was investigated by time-resolved SAXS experiments at the ESRF. The kinetic pathway of the micellisation was observed for the first time and could be modelled by a nucleation and growth process.
Share
The self-assembly of amphiphilic molecules is at the origin of many fascinating features of soft matter [1]. Observing how these molecules spontaneously order into nanostructures such as micelles and vesicles is an exceedingly difficult task. Controlled self-assembly is essential for the formation of the various components responsible for life as exemplified by living cells. In material science and physical chemistry, self-assembly is an important tool where the manipulation and control of such complex assemblies constitutes a novel route to the design of nanostructures. In order to fully comprehend and exploit such self-assembled structures, the pathways of their formation need to be understood. So far, such studies have been limited because of the lack of both experimental techniques to capture and resolve this very fast process [2] and well-characterised model systems to reproduce the process in a controllable manner [3]. In this study, we have taken advantage of the combination of stopped-flow rapid mixing and time-resolved small-angle X-ray scattering (TR-SAXS) [4]. The amphiphilic block copolymer poly(ethylene-alt-propylene)-poly(ethylene oxide) served as a model system [3] and the spontaneous micellisation was induced by rapid mixing of the block copolymer solution in dimethylformamide (DMF), with a water and DMF mixture which has strong selectivity towards the poly(ethylene oxide). For the first time we were able to capture and describe how the self-assembly processes take place in real time.
 |
|
Figure 1. Schematic view of the experimental setup used for the stopped-flow time-resolved SAXS experiment. The block copolymer solution in DMF is diluted by water to initiate the self-assembly process. td is the dead time between the rapid turbulent mixing and the first observable point (< 4 milliseconds) during which a homogenous polymer concentration on the molecular scale is reached by diffusion. The inset shows the sequential acquisition scheme used to fill the read out time (tr) of the CCD detector (FRELON). ta and td are the acquisition time and a predefined delay time, respectively. |
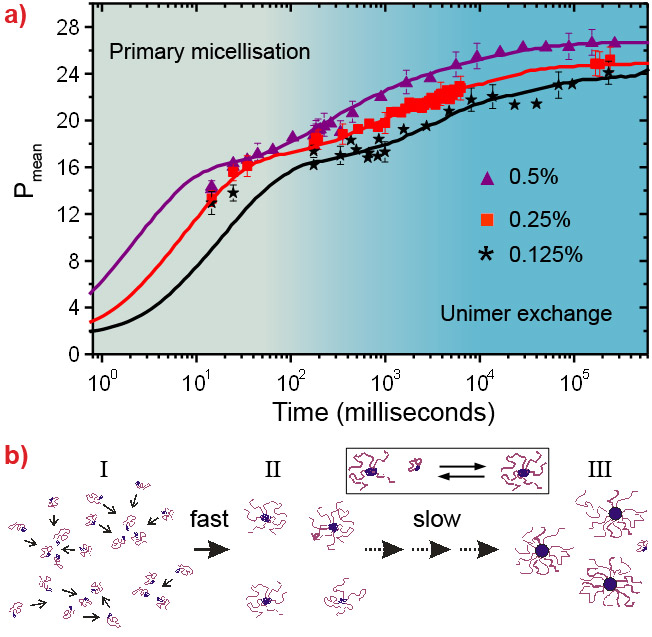
Figure 1 depicts the stopped-flow device coupled to TR-SAXS at beamline ID02 [4]. The stopped-flow device consists of two motorised syringes and a mixer. The block copolymer solution in DMF is rapidly mixed with the water/DMF solution and transferred to the X-ray capillary cell within about 4 ms. The time evolution (tag) of the nanostructure was probed by SAXS employing a sequential data acquisition scheme depicted in the inset. By modelling the time-resolved SAXS patterns by a core-corona structure, we obtained the mean radius of micelles and their aggregation number or the average number of block copolymers per micelle (Pmean), radial density profile, etc. Figure 2a shows the time evolution of Pmean, demonstrating an intrinsically broad growth process which cannot be fully described by a simple relation such as the sum of two exponential functions. The data at the shortest times suggest the existence of a fast initial aggregation. This process seems to become exhausted at intermediate times leading to a shoulder of Pmean that changes with concentration. The terminal relaxation towards the final equilibrium then appears to slow down with time - the overall rate increasing with concentration. The observed behaviour can be quantitatively reproduced using a kinetic model involving insertion and expulsion of single block polymer chains (unimers) within the classical nucleation and growth theory [5]. According to this theory, the growth of the micelles is regulated by a micellisation potential that is essentially given by the difference of the free energy of a micelle and an equivalent amount of unimers properly taking into account the translational entropy. The prediction in Figure 2a corresponds to an effective microscopic interfacial tension which is close to the known macroscopic interfacial tension for the block copolymer in these solvent mixtures. In this model, the initial free unimers are consumed rapidly in the nucleation leading to metastable micelles (region I). These micelles can further grow only if other micelles dissolve by releasing unimers. This is particularly clear from the micellar size, represented by Pmean, exhibiting a shoulder at the time when the unimers are consumed below their critical micellar concentration level (region II). Interestingly, this shoulder is also characterised by a peak in the width of the Gaussian micellar distribution function (data not shown). The broader distribution signifies micellar rearrangement processes where small micelles shrink and large micelles grow. At later times, the distribution narrows again as the final equilibrium is approached (region III). This scenario is presented schematically in Figure 2b.
 |
|
Figure 2. a) Time evolution of the mean aggregation number of micelles (Pmean) for different concentrations of the block copolymer. Continuous lines represent fits to the nucleation and growth model. b) Schematic representation of the micellisation process involving a fast nucleation in which the unimer concentration is depleted and a slow growth to micelle/unimer equilibrium. |
In summary, the kinetics of the formation of block copolymer micelles has been directly observed in situ by synchrotron SAXS with millisecond time resolution. The formation and growth of micelles can be quantitatively described by a nucleation and growth type process governed by the elemental unimer insertion or expulsion mechanism. The kinetic stability and the growth rate are intimately controlled by a system specific micellisation potential which can be deduced from structural and thermodynamic investigations. These findings open a new road to a predictive scheme for the aggregation of surfactant/block copolymer molecules into nanoparticles [6]. The final properties (equilibrium or nonequilibrium) can be controlled not only by the chemical nature and sizes of the copolymer blocks, but also by simple physical parameters such as the polymer concentration and interfacial tension. By using this knowledge, one may envisage new ways of designing and manipulating nanostructures in an intelligent and predictive way.
References
[1] P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers (Elsevier, Amsterdam, 2000).
[2] T.M. Weiss, T. Narayanan, C. Wolf, M. Gradzielski, P. Panine, S. Finet, and W. Helsby, Phys. Rev. Lett. 94, 038303 (2005).
[3] R. Lund, L. Willner, J. Stellbrink, P. Lindner, and D. Richter, Phys. Rev. Lett. 96, 068302 (2006).
[4] P. Panine, S. Finet, T. Weiss, and T. Narayanan, Adv. Colloid Interf. Sci. 127, 9 (2006).
[5] Nucleation Theory and Applications, J.W.P. Schmelzer (Wiley-VCH Verlag GmbH & Co., KGaA, Weinheim, 2005).
[6] X. He and F. Schmid, Phys. Rev. Lett. 100, 137802 (2008).
Principal publication and authors
R. Lund (a), L. Willner (b), M. Monkenbusch (b), P. Panine (c), T. Narayanan (c), J. Colmenero (a), and D. Richter (b), Structural Observation and Kinetic Pathway in the Formation of Polymeric Micelles, Physical Review Letters 102, 188301 (2009).
(a) Donostia International Physics Center/University of the Basque, Donostia-San Sebastián (Spain)
(b) Institute for Solid State Research, Forschungszentrum Jülich (Germany)
(c) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.