- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2016
- Matter at extremes
- A reversible Ru-Cs particle reconstruction demonstrates the role of Cs promotion in the Fischer-Tropsch reaction
A reversible Ru-Cs particle reconstruction demonstrates the role of Cs promotion in the Fischer-Tropsch reaction
Time-resolved X-ray absorption studies of the Ru-Cs system under in situ conditions reveal a reversible particle reconstruction upon syngas reactant switching. This work unravels Fischer-Tropsch gas-phase component interaction with the active metal Ru mediated by Cs promotion and determines the characteristics of the metal and the promoter.
The Fischer–Tropsch (FT) synthesis allows the transformation of syngas, a mixture of CO and H2, into clean hydrocarbons that are convenient substitutes for compounds produced by oil refining. Ruthenium catalysts present some unique features in FT synthesis as they possess higher intrinsic activity than other metals and can work under higher partial pressures of water or other oxygenate-containing atmospheres. Such atmospheres are particularly important when using biomass produced syngas.
FT catalysts often contain chemical promoters such as alkali elements. An alkali metal increases both the activity and selectivity for high molecular weight hydrocarbons and also favours the formation of olefinic products [1]. This enhanced activity/selectivity is often explained in terms of the basicity of the alkali promoter, which is suggested to increase the CO dissociative adsorption rate, resulting in an increase in the surface coverage of dissociated CO species. This results in higher selectivities towards longer chain hydrocarbon and olefin products [1]. Some interpretations state that the alkali element is an electronic promoter which donates a part of its electron density to Ru even in its oxidised state. Also, another debate is ongoing about the alkali promoter location in the catalyst, which could be inside or at the surface of the metal nanoparticles, or segregated on the support surface with the promotion occurring at the contact point between metal and alkali nanoparticles [2].
In this work, high surface area graphite carbon was employed due to its surface inertness that could facilitate the reduction of the metal precursor to the zero valence state and favour the metal-promoter interaction over that of the metal-support [2]. Furthermore, this support presents good stability for metal particles due its low reactivity to gasification under reaction conditions and high external surface area without micropores which precludes mass transfer problems during the reaction.
Specially-designed time-resolved X-ray absorption spectroscopy experiments [3], performed at beamline ID24 under temperature-programmed reduction (TPR) conditions and during FT reactant (H2/CO) switching have allowed us to unravel Fischer-Tropsch gas-phase component interaction with the active metal Ru mediated by Cs promotion and to determine the electronic properties and local chemical environment of the metal and the promoter supported on graphite materials.
 |
|
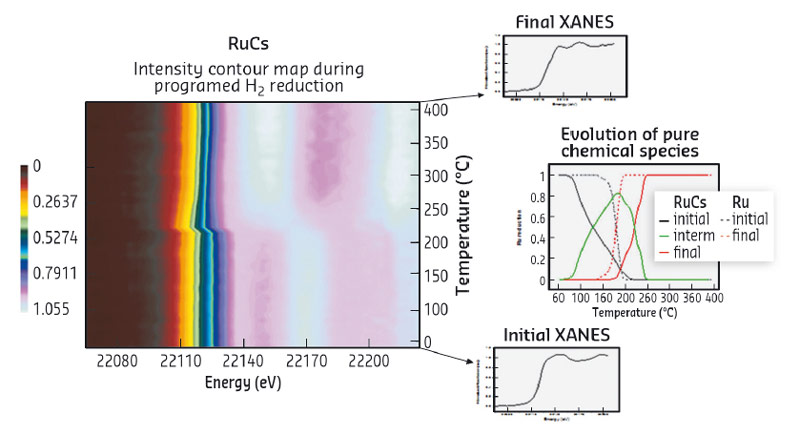
Fig. 120: Normalised absorbance intensity contour map during programmed H2 reduction for 4%Ru-4%Cs/C. XANES spectra and concentration evolution of the pure chemical species present along H2-TPR. |
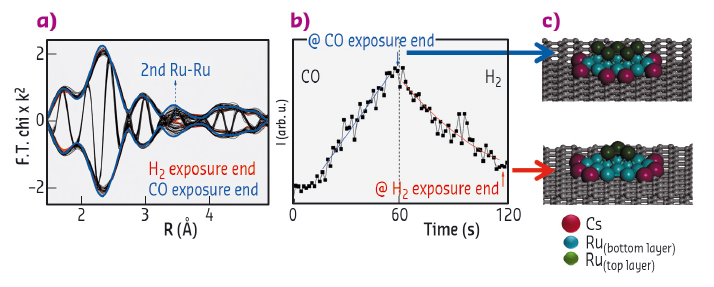
XANES analysis at both Ru K- and Cs L1-edges was carried out during TPR conditions. This allowed us to learn that Ru reduction is a complex process occurring in two steps via an intermediate oxidation state and to show the concomitant Cs partial reduction (Figure 120). Time resolved EXAFS analysis at Ru K-edge proved the presence of Cs atoms as first neighbours of Ru. Fitting the data gave structural parameters which match with Ru-Cs nanoparticles where the Cs atoms surround the Ru cluster. Under H2 atmosphere, a new feature around 3 Å (at close distance from the Ru-Ru first shell) was revealed and this can be attributed to Cs atoms in the vicinity of Ru entities. Moreover, the near absence of the second Ru-Ru coordination shell in H2 suggests a particle morphology resembling a flat single-layer structure. However, when the H2 atmosphere is switched to CO, the appearance and gradual increase of Ru neighbours in the second shell implies some restructuring of the nanoparticles to a 3D shape with low first and second shell coordination numbers. In addition, the Ru-Cs particle morphology changes reversibly during cycles of alternate H2/CO exposure, staging the strong interaction of CO with the surface of the Ru-Cs nanoparticles supported on graphite (Figure 121). The latter has been measured directly by microcalorimetry of CO adsorption.
 |
|
Fig. 121: a) Modulus and imaginary part Fourier transform of EXAFS spectra obtained during a H2 to CO switch. b) 2nd Ru-Ru coordination shell time evolution during one CO-H2 cycle for 4%Ru4%Cs/C catalyst. c) Calculated morphologies for the Ru nanoparticles after CO or H2 exposure, respectively. |
Therefore, this study provides evidence of the electronic interaction between Cs and Ru. The presence of the alkali leads to a stronger interaction with the CO molecule. This in turn results in an increase of the adsorbed CO coverage and likely hinders the hydrogen adsorption, resulting in higher olefin selectivity and more long-chain hydrocarbon production in the Fischer-Trospch reaction.
Principal publication and authors
Time resolved XAS investigation of the local environment and evolution of oxidation states of a Fischer-Tropsch Ru-Cs/C catalyst, J.L. Eslava (a), A. Iglesias-Juez (a), G. Agostini (b), M. Fernández-García (a,c), A. Guerrero-Ruiz (c,d) and I. Rodríguez-Ramos (a,c), ACS Catal. 6, 1437-1448 (2016); doi: 10.1021/acscatal.5b02489.
(a) Instituto de Catálisis y Petroleoquímica, CSIC, Madrid (Spain)
(b) ESRF
(c) Grupo de Diseño y Aplicación de Catalizadores Heterogéneos, Unidad Asociada UNED-CSIC (ICP) (Spain)
(d) Departamento de Química Inorgánica y Química Técnica, UNED, Madrid (Spain)
References
[1] N. Lohitharn et al., J. Catal. 260, 7-16 (2008).
[2] F. R. García-García et al., Phys. Chem. Chem. Phys. 13, 12892-12899 (2011).
[3] A. Iglesias-Juez et al., J. Amer. Chem. Soc. 133, 4484-4489 (2011).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.