- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2002
- Surface and Interface Science
- Following Adsorption Kinetics at Electrolyte/Metal Interfaces Through Crystal Truncation Scattering: Sulphur on Au(111)
Following Adsorption Kinetics at Electrolyte/Metal Interfaces Through Crystal Truncation Scattering: Sulphur on Au(111)
The interaction of sulphur (S) and S-headed organic molecules with metallic surfaces is of interest since S is a poison in heterogeneous catalysis, and S-headed organic molecules, such as alkanethiols, form self-assembled monolayers which could play a role in nanotechnology. The adsorption of these species on Au(111) has been taken as a model system for interfacial science research. In particular, adsorption from aqueous solutions deserves special interest since many technological processes take place in such an environment. We have made combined in situ scanning-tunnelling microscopy (STM), and surface X-ray diffraction (SXRD) studies under electrochemical control for S and S-headed organic molecules on Au(111) at beamline ID32.
 |
|
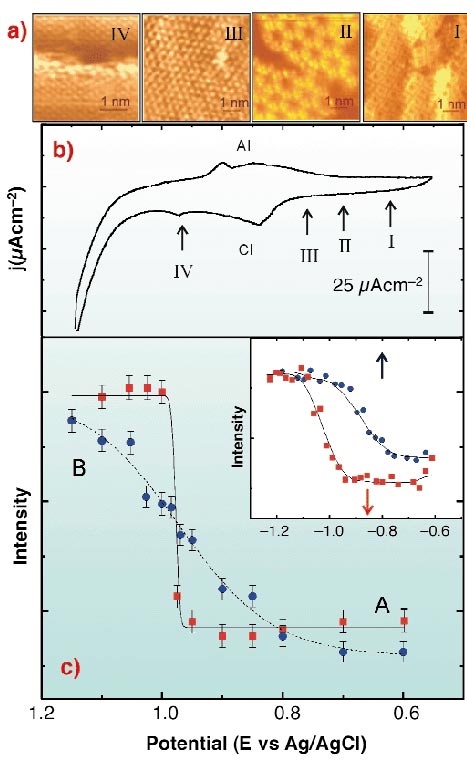
Fig. 41: (a) In situ STM images taken at the potentials indicated in (b). (I) S8; (II) S3 forming a honeycomb array; (III) S ( |
 ) and positive (
) and positive ( ) scan directions. Lines are fitted to the data as a guide for the eye. Inset: the same for hexanethiol/Au(111) in 0.1 M NaOH. Red and blue arrows indicate the position of the electrodesorption and electroadsorption peaks potentials from the voltamogram, respectively.
) scan directions. Lines are fitted to the data as a guide for the eye. Inset: the same for hexanethiol/Au(111) in 0.1 M NaOH. Red and blue arrows indicate the position of the electrodesorption and electroadsorption peaks potentials from the voltamogram, respectively.In situ STM imaging reveals different S species depending on the potential (E) applied to the Au(111)/electrolyte interface (Figure 41a). When E is shifted from -0.6 V to 0.8 V (Figure 41b), S desorption takes place according to the following pathway: S8 ![]() S3
S3 ![]() monomeric S in (
monomeric S in ( 3x3)R30° [1]. In this potential range (3x3)R30° S, S3 and S8 structures can coexist due to slow desorption kinetics. When E is scanned across peak CI (at -0.84 V) the following reaction could take place:
3x3)R30° [1]. In this potential range (3x3)R30° S, S3 and S8 structures can coexist due to slow desorption kinetics. When E is scanned across peak CI (at -0.84 V) the following reaction could take place:
2e- + S-Au(111) + H2O = SH- + OH- + Au(111)
2e- + S-Au(111) = SH2- + Au(111)
Thus, the (3x3)R30° S lattice is no longer observed, and only the Au(1x1) lattice and S at steps are detected. Potential scan reversal results in S adsorption through the opposite pathway, leading finally to S8 structures.
The surface structure involves small domains of different sulphur species that arrange in different lattices (Figure 41). This precludes the possibility of observing fractional order diffracted beams. We have performed in situ  -scans for different E at L = 2.5 for the (10) crystal- truncation rod (CTR), and simultaneously recorded current density (j) vs. E profiles. We have proved by simulations that the integrated intensity in anti-phase conditions of this CTR is sensitive to the total amount of S in the surface, and therefore, has been used to follow changes in the amount of adsorbed species induced by changes in E, i.e. a cyclic diffractogram (CD). Two well-defined regions of almost constant diffraction intensity, separated by a sudden rise at E = -0.98 V can be observed, corresponding to potentials where STM shows the presence (A) or absence (B) of adsorbed S on Au(111) terraces (Figure 41c). By repetitive cycling the CD shows that S adsorption/desorption is completely reversible. The fact that the sudden rise in the diffraction intensity takes place at -0.98 V, i.e. at E values where chemisorbed S has already been completely transformed into sulphide, suggests that sulphide remains on the Au(111) surface in a physisorbed state. During sulphide re-adsorption diffraction intensity decays to a minimum well before chemisorption (which takes place at AI), also indicating a physisorption step. The discontinuity in the negative scan of the CD strongly suggests that desorption of the physisorbed layer involves a first order phase transition. Contrary to this, the weak adsorption of the sulphide species from solution increases smoothly with E and the shape of the diffraction intensity plot fits a Langmuir isotherm reasonably well. We have obtained similar results for the hexanethiol/Au(111) interface (inset in Figure 41c): a physisorbed hexanethiolate state is identified. The fact that S and alkanethiolate exhibit the same trend demonstrates that the physisorbed state is mediated by the S head, rather than by hydrocarbon chains, which would only contribute to its stabilisation [2].
-scans for different E at L = 2.5 for the (10) crystal- truncation rod (CTR), and simultaneously recorded current density (j) vs. E profiles. We have proved by simulations that the integrated intensity in anti-phase conditions of this CTR is sensitive to the total amount of S in the surface, and therefore, has been used to follow changes in the amount of adsorbed species induced by changes in E, i.e. a cyclic diffractogram (CD). Two well-defined regions of almost constant diffraction intensity, separated by a sudden rise at E = -0.98 V can be observed, corresponding to potentials where STM shows the presence (A) or absence (B) of adsorbed S on Au(111) terraces (Figure 41c). By repetitive cycling the CD shows that S adsorption/desorption is completely reversible. The fact that the sudden rise in the diffraction intensity takes place at -0.98 V, i.e. at E values where chemisorbed S has already been completely transformed into sulphide, suggests that sulphide remains on the Au(111) surface in a physisorbed state. During sulphide re-adsorption diffraction intensity decays to a minimum well before chemisorption (which takes place at AI), also indicating a physisorption step. The discontinuity in the negative scan of the CD strongly suggests that desorption of the physisorbed layer involves a first order phase transition. Contrary to this, the weak adsorption of the sulphide species from solution increases smoothly with E and the shape of the diffraction intensity plot fits a Langmuir isotherm reasonably well. We have obtained similar results for the hexanethiol/Au(111) interface (inset in Figure 41c): a physisorbed hexanethiolate state is identified. The fact that S and alkanethiolate exhibit the same trend demonstrates that the physisorbed state is mediated by the S head, rather than by hydrocarbon chains, which would only contribute to its stabilisation [2].
In conclusion, the combination of CD, STM and electrochemistry is a powerful tool for the detection of different adsorbed states. It allows us to obtain information of adsorption kinetics for both relatively simple systems and highly-reactive and complex systems such as S and alkanethiols, respectively on Au(111).
References
[1] C. Vericat, G. Andreasen, M. E. Vela, R. C. Salvarezza, J. Phys. Chem. B 104, 302 (2000).
[2] C. Vericat, G. Andreasen, M. E. Vela, H. Martin, R. C. Salvarezza, J. Chem. Phys. 115, 6672 (2001).
Principal Publication and Authors
C. Vericat (a), M. E. Vela (a), G. A. Andreasen (a), R. C. Salvarezza (a), F. Borgatti (b), R. Felici (b), T.-L. Lee (c), F. Renner (c), J. Zegenhagen (c), J.A. Martín-Gago (d), Phys. Rev. Lett., in press.
(a) INIFTA (Argentina)
(b) INFM-OGG c/o ESRF
(c) ESRF
(d) Instituto Ciencia de Materiales de Madrid-CSIC (Spain)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.