- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2003
- Soft Condensed Matter
- Deformation Studies of Single PBO Fibres
Deformation Studies of Single PBO Fibres
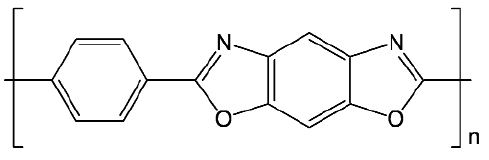
Poly(p-phenylene benzobisoxazole) or PBO fibres are so called 'high-performance' polymer fibres which are amongst a group of materials known as 'rigid-rods'. Their molecular backbone repeat unit has a rigid structure (Figure 78) which contributes towards the fibres' remarkably high mechanical properties, typically more than double those of other high performance fibres such as poly(p-phenylene terephthalamide) or PPTA (Kevlar®) [1].
 |
|
Fig. 78: Chain repeat unit of PBO. |
Three varieties of PBO fibre were examined within this study, AS, HM and HM+. All three were manufactured by Toyobo (Japan), using a dry-jet wet-spinning process [2]. The AS fibre was provided literally 'as-spun'. The HM and HM+ fibre types were given a heat-treatment process to increase their fibre modulus. This involved the application of tension at high temperature. To further increase the modulus of the HM+ variety, a modified coagulated process was used [2]. The resulting fibre modulus values were 180 GPa, 254 GPa and 330 GPa for PBO AS, HM and HM+ respectively [1].
It is clear from the modulus variations between fibre types that processing parameters are important in determining final mechanical properties. The aim of this experiment was therefore to investigate this influence of the microscopic structure upon the macroscopic mechanical properties. It is well established that crystallite orientation is one of the most critical factors in this relationship. Microdiffraction can readily provide such information and may additionally be used to probe inhomogeneities such as 'skin-core' structural differences in single fibres.
Microdiffraction was carried out on beamline ID13. A scanning set-up was used with a beam spot-size of 3 µm. The fibres were mounted within a deformation device and deformed in situ. At predetermined load levels, diffraction patterns were generated across the fibres at 2 µm intervals. This is shown in Figure 79 where diffraction patterns generated across a single fibre are superimposed on a scanning electron micrograph (scaled for spot-size and position, the vertical offset being for clarity only). Thus, differences in structure between the fibre 'skin' and 'core' may be investigated.
 |
|
Fig. 79: Across-fibre diffraction patterns imposed over a SEM micrograph of a single PBO AS fibre (diffraction patterns scaled for beam-size and position). |
Crystallite orientation was calculated from azimuthal broadening of the (200) equatorial reflection. The <sin2![]() > orientation parameter was used, ranging from <sin2
> orientation parameter was used, ranging from <sin2![]() > = 0.667 for an un-oriented structure and <sin2q> = 0 for perfect crystallite orientation.
> = 0.667 for an un-oriented structure and <sin2q> = 0 for perfect crystallite orientation.
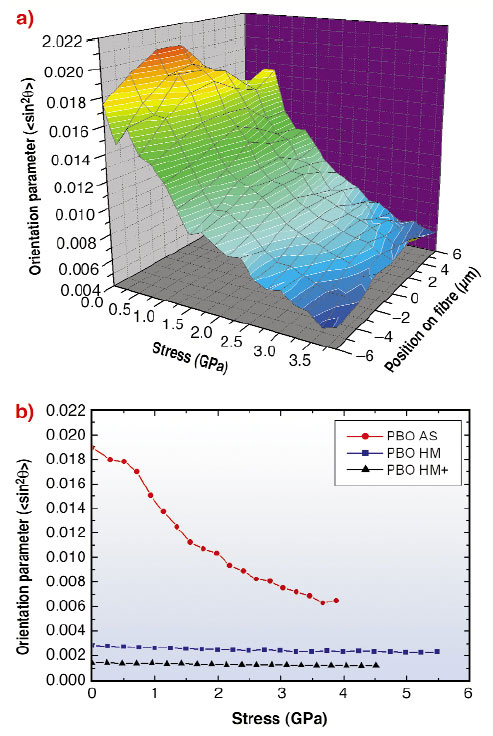
Figure 80a shows the change in orientation parameter across a single fibre (in this case PBO AS) with respect to stress. Initially (prior to loading), the fibre 'core' has a lower degree of crystallite orientation than the 'skin'. This can be attributed to shear forces generated during the spinning process. This 'skin-core' structural difference was only found in the AS fibre type indicating that heat-treatment increases fibre structure homogeneity. Additionally, the degree of crystallite orientation increases with increasing stress at all positions across the fibre.
 |
|
Fig. 80: (a) Changes in crystallite orientation parameter across a single PBO AS fibre with applied stress and (b) weighted-average crystallite orientation with applied stress for PBO AS, HM and HM+. |
Figure 80b shows differences in the weighted-average orientation parameter with stress for all fibre types. The PBO AS fibre has the lowest degree of crystallite orientation and the greatest increase in orientation with applied stress. Both the HM and HM+ fibre types show a significantly higher degree of orientation and less change in orientation with stress. It is clear that both the degree of crystallite orientation and orientation change with stress is in the same fibre order as tensile modulus.
In conclusion, the microbeam scanning setup at ID13 offers an ideal means of investigating structural differences in single fibres. The addition of an in situ deformation device enables simultaneous generation of tensile and crystallographic structural information. The results presented here demonstrate that crystallite orientation is one of the most important structural factors in determining fibre tensile modulus. This can be attributed to the fact that increasing crystallite orientation increases the influence of direct chain stretching during deformation.
References
[1] R.J. Davies, M.A. Montes-Morán, C. Riekel and R.J. Young, J. Mater. Sci. 36, 3079 (2001).
[2] T. Kitagawa, M. Ishitobi and K. Yabuki, J. Polym. Sci.; Polym. Phys. Ed. 38, 1605 (2000).
Principal Publication and Authors
R.J. Davies (a), M.A. Montes-Morán (a), C. Riekel (b), R.J. Young (a), J. Mater. Sci. 38, 2105 (2003).
(a) UMIST/University of Manchester (UK)
(b) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.