- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2006
- Soft Condensed Matter
- Simultaneous micro-Raman spectroscopy and microfocus X-ray diffraction
Simultaneous micro-Raman spectroscopy and microfocus X-ray diffraction
Raman spectroscopy and X-ray scattering are complementary techniques. Whilst one provides information relating to molecular bond energies and orientations, the other probes the sample’s crystallographic structure and morphology. Not only do they provide differing information, but this relates to quite different length scales within a material’s hierarchical structure. However, the two techniques also share several similarities. For example, they are non-contact and non-destructive for many materials, and neither requires modified or coated samples. Consequently both are used extensively for in situ studies such as investigating the effects of deformation, temperature or pressure.
During recent years, Raman spectroscopy and X-ray scattering have independently evolved microfocus capabilities. With smaller laser and X-ray beams, smaller structural heterogeneities can be resolved from within a single sample. Microfocussing also allows confined sample volumes to be studied, and reduces averaging when performing in situ experiments (such as chemical modification). The combination of µRaman and µXRD on the ID13 beamline provides a completely new tool for the characterisation of materials [1]. Whilst the techniques’ similarities make their combination practical, their differences make it scientifically important. A combinatorial approach also ensures a common sampling location on the specimen, consistency during dynamic studies, reduces experimental time, and is beneficial for rare and valuable samples (which may be only available in small volumes).
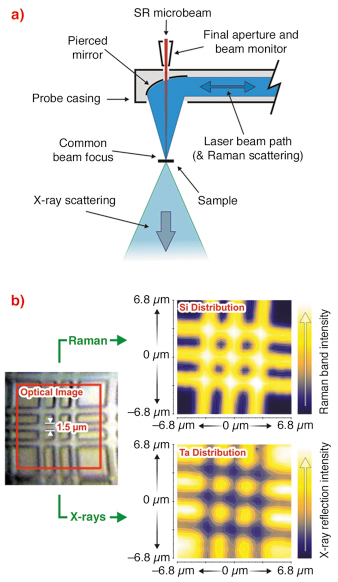
The ID13 beamline’s µRaman apparatus was designed in collaboration with Renishaw. It comprises an Invia spectrometer and 785 nm near-IR laser, coupled via a single-mode fibre optic to a remote probe located within the sample environment. The spectrometer has a 1200 lines/mm grating whilst the probe head incorporates an integrated camera and white-light illumination for sample (and beam) visualisation. The system is designed in an on-axis configuration with the laser beam delivered to the same point on the sample as the X-ray beam, simultaneously. This is made possible by a pierced mirror within the probe head which focuses the laser beam onto the sample whilst allowing the X-ray beam to pass through (Figure 52a). The laser beam spot size at the focal position is approximately 1 µm. This is comparable to the X-ray beam size from a number of different ID13 optics [2]. Both techniques can therefore resolve microstructural variations, such as 1.5 µm Si/Ta structures on laboratory-produced samples (Figure 52b).
 |
|
Fig. 52: a) Schematic showing on-axis beam delivery within the probe head and b) a Si/Ta structured sample from which simultaneous Raman and X-ray scattering reveals the spatial distribution of Si and Ta independently. |
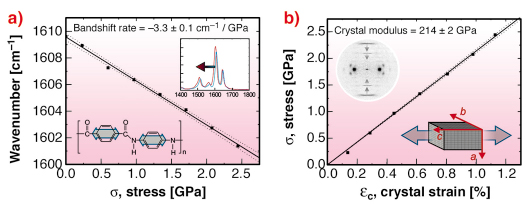
In a recent demonstration of the system’s capabilities, a single poly(p-phenylene terephthalamide), PPTA or Kevlar fibre was deformed in situ. Raman spectra and diffraction patterns were recorded at regular intervals using simultaneous exposures of 5 seconds. The Raman spectra of PPTA is characterised by a strong band at 1610 cm-1. This is assigned to a vibrational mode of the p-phenylene ring and shifts to a lower wavenumber under tension. This band shift corresponds to deformation of the fibres molecular “backbone”. The diffraction pattern of PPTA has several meridional layer lines corresponding to the axial crystal lattice spacing (c-spacing). Changes in layer line position under tension reflect direct distortion of the crystal lattice (crystal strain). During deformation, both the 1610 cm-1 Raman band and crystal strain change linearly with macroscopic stress (Figure 53). This indicates stress transfer between crystalline domains and individual molecular bonds. The resulting Raman band shift rate and crystal modulus are -3.3 cm-1/GPa and 214 GPa respectively. Deformation also causes a stress-induced increase in the degree of crystalline domain orientation consistent with the non-reversible rotation of chains.
 |
|
Fig. 53: a) Variation of the 1610 cm-1 Raman band and b) crystal strain with applied stress (determined simultaneously). Inset graphics reflect how the Raman band and layer line shifts correspond to p-phenylene ring stretching and axial lattice distortion respectively. |
Whilst the PPTA diffraction pattern provides information exclusive to the crystalline phase, its Raman spectra contains both a crystalline and amorphous contribution. Combining this information allows individual contributions to be isolated. For example, broadening of the 1610 cm-1 Raman band during deformation suggests an increasing stress heterogeneity between the crystalline and amorphous fractions. This can be explained in terms of low- and high-stress uniform-strain mechanical models. At low stresses, linear Raman band broadening can be attributed to a phase-related difference in mechanical properties. At high stresses, an increasing degree of crystalline domain orientation causes heterogeneous stiffening within the PPTA fibre’s structure. Consequently, the amorphous fraction remains “under-stressed” during subsequent deformation.
Combining µRaman and µXRD has provided a new insight into deformation micromechanics within single PPTA fibres. It reveals how molecular- and crystallographic-scale contributions influence a material’s mechanical properties and how heterogeneous stiffening can occur within a material’s microstructure.
References
[1] R.J. Davies, M. Burghammer, C. Riekel, Applied. Physics Letters, 87(26), 264105 (2005).
[2] C. Riekel, R.J. Davies, Current Opinion in Colloid & Interface Science, 9, 396–403 (2005).
ESRF
Principal Publication and Authors
R.J. Davies, M. Burghammer, C. Riekel, Macromolecules 39, 4834-4840 (2006).
ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.