- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2013
- Electronic structure and magnetism
- Preference towards five-coordination in Ti silicalite-1 upon molecular adsorption
Preference towards five-coordination in Ti silicalite-1 upon molecular adsorption
Meso- and micro-porous Ti-zeotype materials are often good catalysts for selective oxidation reactions and thus are of interest for the petrochemical industry. One of the most extensively used is titanium silicalite-1 (TS-1) [1,2]. Analysing the Ti K-edge extended X-ray absorption fine structure (EXAFS), it has been established that Ti is preferentially located inside a tetrapodal structure within the silicalite framework. EXAFS has also been used to investigate the catalyst under in situ conditions and to understand the mechanism of the catalytic transformations using water and ammonia as probe molecules due to their importance in various catalytic transformations [1,2]. However, the EXAFS data were not conclusive and other techniques such as microcalorimetric measurements have been employed. It was proposed that the Ti coordination number is close to six upon adsorption of water and ammonia [1], i.e. two adsorbed molecules on Ti. But these molecules can also adsorb on the zeolite-framework, therefore the number of adsorbed molecules per Ti centre was obtained by subtracting the same signal obtained on a Ti-free silicalite-1. Unfortunately, the reliability of this method is under debate.
To address this problem we have performed a valence-to-core X-ray emission spectroscopic (vtc-XES) investigation on TS-1 exposed to ammonia (50 torr) and water (vapour pressure) at beamline ID26. vtc-XES allows the valence electronic levels just below the Fermi energy to be probed and thus is a valuable tool for studying the chemical bond between metal ion and ligands [3]. We have modelled the experimental spectra using density functional theory (DFT) calculations based on Ti-centred clusters that mimic the environment of Ti in the activated catalyst (TS-1/act), in interaction with water (TS-1/H2O) and ammonia (TS-1/NH3) [3]. One or two molecules of water/ammonia can adsorb on the Ti ion and thus we have built clusters with one (calc1) and two (calc2) molecules directly bonded to the metal centre.
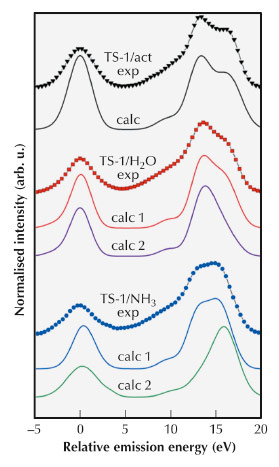
Figure 93 shows the vtc-XES of TS-1/act, TS-1/H2O and TS-1/NH3 in comparison with the theoretical spectra computed using the ORCA package.
 |
|
Fig. 93: Experimental (exp) and calculated (calc) vtc-XES spectra of TS-1/act, TS-1/H2O and TS-1/NH3. In both cases we calculated two spectra with one (calc 1) and two (calc 2) adsorbed molecules. The vtc-XES spectra are offset along the intensity axis for clarity. |
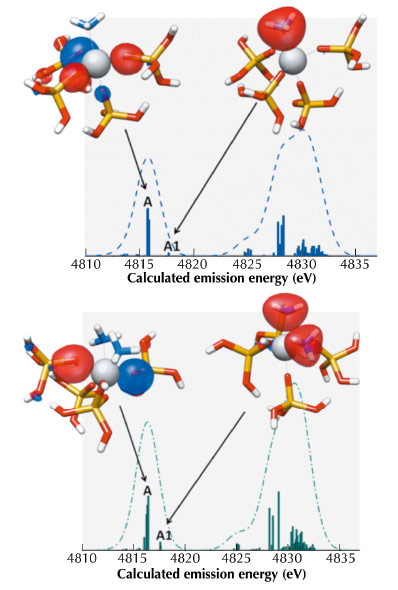
We observe that the theoretical vtc-XES of TS-1/act, TS-1/H2O (calc1) and TS-1/NH3 (calc1) present similar relative changes in intensity as the experimental vtc-XES spectra. When one molecule is adsorbed on the Ti centres, the degeneracy of the molecular orbitals (MOs) linked to the main features of the vtc-XES (see Figures 93 and 94) is removed and new transitions involving MOs with Owater(2p) and Nammonia(2p) atomic character arise in the Kβ2,5 region [3]. The calculation reproduces the shift of the feature A (Kβ’’ emission lines), which we attribute to the presence of transitions involving MOs with Owater(2s) and Nammonia(2s) atomic character.
 |
|
Fig. 94: vtc-XES of two clusters of TS-1 with one (calc1, top) and two (calc2, bottom) molecules of ammonia adsorbed. The molecular orbitals that affect the emission line A (i.e., Kβ”) are also reported. The colour code is grey for Ti, red for O, blue for N and yellow for Si. |
In summary, we find strong experimental evidence that only one molecule is adsorbed on the Ti centres in TS-1. Knowledge of the number of molecules coordinated to the Ti centres in TS-1 under in situ conditions is of paramount importance for understanding the catalytic path of the catalyst and provides crucial input for theoretical estimates of efficiency and kinetics of a reaction. Our conclusion mainly relies on the comparison between experiment and quantum chemical calculations. The recent progress in modelling of XES data using density functional theory makes this a viable approach that allows a detailed understanding of the underlying modifications of the electronic structure.
Principal publication and authors
E. Gallo (a), F. Bonino (b), J.C. Swarbrick (a), T. Petrenko (c), A. Piovano (d) S. Bordiga (b), D. Gianolio (e), E. Groppo (b), F. Neese (c), C. Lamberti (b) and P. Glatzel (a), ChemPhysChem 14, 79–83 (2013).
(a) ESRF, Grenoble CEDEX (France)
(b) Department Chemistry, University of Turin (Italy)
(c) Max-Planck Institute for Bioinorganic Chemistry, Mülheim (Germany)
(d) ILL, Grenoble (France) (e) Diamond Light Source Ltd., Didcot (UK)
References
[1] S. Bordiga, F. Bonino, A. Damin and C. Lamberti, Phys. Chem. Chem. Phys. 9, 4854-4878 (2007).
[2] S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev. 113, 1736-1850 (2013).
[3] E. Gallo, C. Lamberti and P. Glatzel, Phys. Chem. Chem. Phys. 13, 19409-19419 (2011).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.