- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2013
- Structural biology
- Solution structure of the hepatitis C virus IRES: an articulated RNA molecule involved in cap-independent translation initiation
Solution structure of the hepatitis C virus IRES: an articulated RNA molecule involved in cap-independent translation initiation
Hepatitis C virus (HCV) infects over 170 million people worldwide, causing chronic liver diseases that can progress to cirrhosis and cancer, and kills more than 350,000 people every year. While recent advances in treatment have been made, there is no vaccine, and HCV remains a significant human health problem. The HCV genome is a positive-sense 9.6 kilobase RNA containing highly structured 5’ and 3’ untranslated regions (UTR). The 5’ UTR comprises an internal ribosome entry site (IRES) encompassing nucleotides 40 to 341, which directs the cap-independent recruitment of both ribosomal subunits, the assembly of a functional translation initiation complex and the translation of a single precursor polyprotein, which is subsequently processed by viral and cellular proteases.
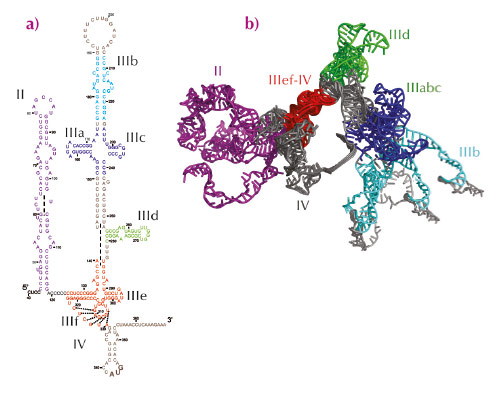
The HCV IRES is a large RNA region comprising a pseudoknot and three structural domains: two long stem loops (domains II and III) and a spur (domain IV) (Figure 56a). Two regions, the IIIabc four-way junction and the IIIef/IV pseudoknot, involve tertiary contacts, and the entire IRES adopts a defined ion-dependent fold under physiological conditions, although it does not form a compact, globular structure. Because the HCV IRES is essential for viral replication, and its sequence is well conserved among all HCV genotypes, its structure may represent a novel target for drug design. In recent years, atomic structures of different fragments of this RNA region have been determined, but the presence of several hairpin loops and regions predicted to be single-stranded confers dynamic flexibility to the entire IRES molecule that has so far hampered its complete structural characterisation by X-ray crystallography or electron microscopy. The structural information available about the entire molecule has remained limited to cryo-electron microscopy reconstructions of the IRES bound to different partners.
 |
|
Fig. 56: Secondary structure of hepatitis C virus internal ribosome entry site (IRES). a) Predicted secondary structure. b) Ensemble structure that collectively best reproduced the experimental SAXS curve. The regions for which an experimentally determined atomic structure is available are shown in the same colour in both figures. |
An atomic model of full-length hepatitis C virus IRES in solution was constructed on the basis of small-angle X-ray scattering (SAXS) data collected at beamlines ID02 and ID14–3 and molecular dynamics simulations. An initial model was constructed by connecting together the known atomic structures of different fragments and constructing the missing parts in accordance with secondary structure predictions (Figure 56a). Molecular dynamics simulations in explicit solvent (ESMD) and normal mode analysis were used to generate an ensemble of 8,000 different physically realistic conformations accessible to the RNA molecule in solution. This ensemble was searched and the model that best reproduced the experimental SAXS scattering curve was selected. In the selected conformer, domain II and domain III extend in opposite directions along the main axis of the molecule forming a right angle, whereas subdomain IIId and domain IV protrude from each side of the molecule. This model is compatible with footprinting experiments performed with chemical and enzymatic probes [1]. ESMD simulations confirmed the flexibility of this molecule and localised the regions of highest flexibility. Principal component analysis (PCA) performed on the ensemble of physically accessible conformers then revealed collective motions in the molecule. In particular, the dominant motion in solution corresponded to the closure of the molecule, with domains II and III remaining in the same plane, acting like jaws that move towards and away from each other. Further refinement revealed that an ensemble of five or more exchanging conformers reproduces SAXS data better than a single structure suggesting, in combination with results from ESMD and PCA, that the hepatitis C virus IRES is an articulated molecule made of rigid structural elements that move relative to each other around flexible joints (Figure 56b). Finally, comparison with the IRES bound to the eIF3 complex and to the ribosomal 40S subunit indicated that motions along the second principal component involving reorientation of domain II relative to domain III must occur upon binding to its partners.
In conclusion, we determined an ensemble structure of the HCV IRES in solution, which highlighted the articulated nature of this RNA molecule. This work offers insights into the conformational changes occurring in IRES upon formation of the translation initiation complex.
Principal publication and authors
J. Pérard (a), C. Leyrat (a), F. Baudin (a), E. Drouet (a) and M. Jamin (a), Nat. Commun. 4, 1612 (2013).
(a) Unit of Virus Host-Cell Interactions, Université Grenoble Alpes-EMBL-CNRS, Grenoble (France)
References
[1] J.S Kieft, K. Zhou, R. Jubin, M.G. Murray, J.Y. Lau and J.A. Doudna, J. Mol. Biol. 292, 513-529 (1999).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.