- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Complex systems and biomedical sciences
- Oxidation-induced nanoscale reorganisation of Pt electrode surfaces
Oxidation-induced nanoscale reorganisation of Pt electrode surfaces
X-ray scattering was used to elucidate the formation and growth of nanoscale islands on platinum electrodes during cyclic electrochemical oxidation and reduction in acid solutions. Determining this structural evolution is crucial in understanding the factors limiting the performance and longevity of fuel cells that use Pt catalysts.
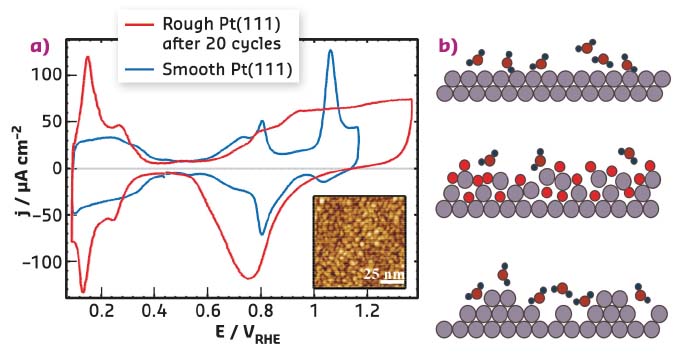
Platinum is by far the most active element for a number of electrocatalytic reactions and it is the catalyst of choice for hydrogen-oxygen fuel cells for vehicle use. A problem for large-scale commercialisation is that the lifetime and performance are partly limited by the properties of the oxide that forms on the surface. The formation and removal of the oxide promotes dissolution and degradation of the catalyst particles. It is well known that repeated cycles of oxidation and oxide reduction lead to irreversible changes in the electrochemical response, which can be attributed to the formation of nanoscale islands on Pt(111) electrode surfaces (Figure 54). However, systematic measurements with surface sensitive techniques, which provide quantitative information on the Pt surface restructuring as a function of electrochemical parameters, were lacking.
 |
|
Fig. 54: a) Electrochemical current-voltage curve (cyclic voltammogram) of pristine, atomically smooth Pt(111) in 0.1 M HClO4 (blue line) and of the same electrode after 20 potential cycles to 1.37 V (red line), showing the irreversible changes in the electrochemical response. The inset shows an STM image of Pt(111) after repetitive cycling. b) Schematic illustration of the structural modification induced by oxidation/oxide reduction (bright red: adsorbed O). |
In previous in situ studies of Pt(111) oxidation in liquid electrolytes by surface X-ray diffraction techniques [1], the locations of the first Pt atoms that move out of their lattice sites in a place-exchange process were determined more precisely than in earlier measurements. Providing the potential is not too high, these atoms can move back to their original locations, but at higher potentials an irreversible restructuring of the surface occurs.
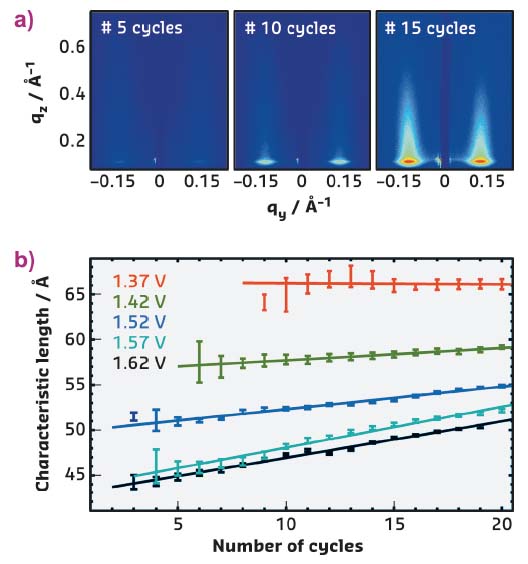
In studies at beamline ID03, the nature of this restructuring was investigated by in situ grazing-incidence small-angle X-ray scattering (GISAXS), which allowed monitoring of the changes in the nanoscale Pt(111) morphology induced by repeated oxidation and reduction cycles. Perchloric acid was chosen as it does not adsorb on the surface, and the number of cycles and the maximum electrode potential were systematically varied. The detector images, recorded in these GISAXS measurements, reveal characteristic diffuse peaks that grow in intensity with increasing number of potential cycles (Figure 55a). The positions and widths of these diffuse peaks provide quantitative information about the size distribution of the nanoscale islands evolving on the surface.
 |
|
Fig. 55: a) In situ GISAXS images show the evolution of diffuse scattering peaks upon repeated cycles to 1.62 V. b) Quantitative studies reveal a gradual evolution of the characteristic lateral dimensions of the nanoscale islands as a function of maximum potential and number of cycles. |
According to these studies, repetitive oxidation and oxide reduction lead to more prominent and homogeneous islands. The characteristic lateral dimensions depend on the upper limit of the cycle and only slightly increase with cycle number (Figure 55b). The structural evolution of the Pt surface morphology strongly resembles that found in studies of Pt(111) homoepitaxial growth and ion erosion in ultra-high vacuum. This can be rationalised by considering that each potential cycle generates a population of Pt adatoms and vacancies on the electrode surface. The GISAXS data suggest that the latter obey the same surface dynamic behaviour as under vacuum conditions, resulting in a similar evolution of the nanoscale morphology.
This remarkable similarity between the microscopic processes during electrochemical Pt(111) oxidation/reduction and those occurring in growth processes at the metal-vacuum interface is not self-evident at all. In many electrochemical systems, surface dynamic processes such as surface diffusion on terraces and across steps, attachment/detachment at steps, and nucleation strongly depend on the potential and co-adsorbed species. Apparently, these effects do not play a significant role for Pt(111) in a non-specifically adsorbing electrolyte such as perchloric acid. This is of key importance for modelling Pt oxidation, restructuring, and dissolution by ab initio theory and molecular dynamics simulations. Here, explicit consideration of the solvent and electrolyte at the electrochemical interface is difficult and currently not feasible for complex processes such as surface transport. The results also suggest that more technologically important cases, such as the oxidation/reduction of Pt nanoparticles, is adequately described without explicit consideration of the electrolyte.
This work shows, for the first time, the detailed quantitative evolution of a Pt surface under oxidation and reduction cycles and it should lead to a better understanding of the dissolution and catalytic degradation processes in fuel cells that are induced by surface oxidation. It also opens up the way for similar studies of oxidation in different electrolytes or of other electrode surfaces as well as for in operando GISAXS studies of the potential-dependent morphology during the oxidation and oxide reduction process itself [2].
Principal publication and authors
Structural reorganization of Pt(111) electrodes by electrochemical oxidation and reduction, M. Ruge (a), J. Drnec (b), B. Rahn (a), F. Reikowski (a), D.A. Harrington (c), F. Carlà (b), R. Felici (b), J. Stettner (a) and O.M. Magnussen (a), J. Am. Chem. Soc. 139, 4532-4539 (2017); doi: 10.1021/jacs.7b01039.
(a) Institut für Experimentelle und Angewandte Physik, Christian-Albrechts-Universität zu Kiel (Germany)
(b) ESRF
(c) Department of Chemistry, University of Victoria (Canada)
References
[1] J. Drnec et al., Electrochim. Acta. 224, 220-227 (2017).
[2] M. Ruge et al., J. Electrochem. Soc. 164, 608-614 (2017).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.