- Home
- News
- Spotlight on Science
- Tracking the protein...
Tracking the protein motions that harvest energy
18-05-2011
Energy conversion is one of the most fundamental processes in all organisms. Glycolysis is the metabolic pathway that breaks down sugars, in the absence of oxygen, to release energy. The pathway is common to all organisms and is very ancient. Indeed, as the pathway does not require oxygen, it is likely to have evolved before the accumulation of oxygen in the earth’s atmosphere. Ten separate enzymes catalyse the different steps in the pathway and the first enzyme to generate energy is phosphoglycerate kinase (PGK). As the enzyme is also a drug target against the causative agents of tetanus and sleeping sickness, and as it activates drugs effective against HIV, understanding its catalytic cycle is important. By combining X-ray crystallography with solution scattering techniques, a high-resolution, time-resolved model of the complete reaction cycle has been established.
Share
Phosphoglycerate kinase (PGK) catalyses the seventh step in glycolysis and is the first enzyme in the pathway to produce energy, rather than consume it, by the creation of adenosine triphosphate (ATP), a molecule that is the universal ‘energy currency’ used for work in cells. As a major controller of flux through the pathway, PGK is a viable target for drugs against anaerobic pathogens, such as Trypanosoma and Plasmodium species (the causative agents of sleeping sickness and malaria), that depend solely on glycolysis for energy metabolism. In addition to its metabolic role, the phosphoryl-transfer activity of PGK is important in the processing of anti-retroviral pro-drugs that take the form of L-nucleoside analogues effective against HIV and hepatitis. The rate-limiting step in the in vivo activation of such compounds is the addition of a third phosphate by PGK. More recently, it has been found that PGK activity is used as a signalling mechanism in oncogenesis. A complete understanding of the functioning of this enzyme is thus essential to improve the processing of pro-drugs and to define its signalling activity in cancer.
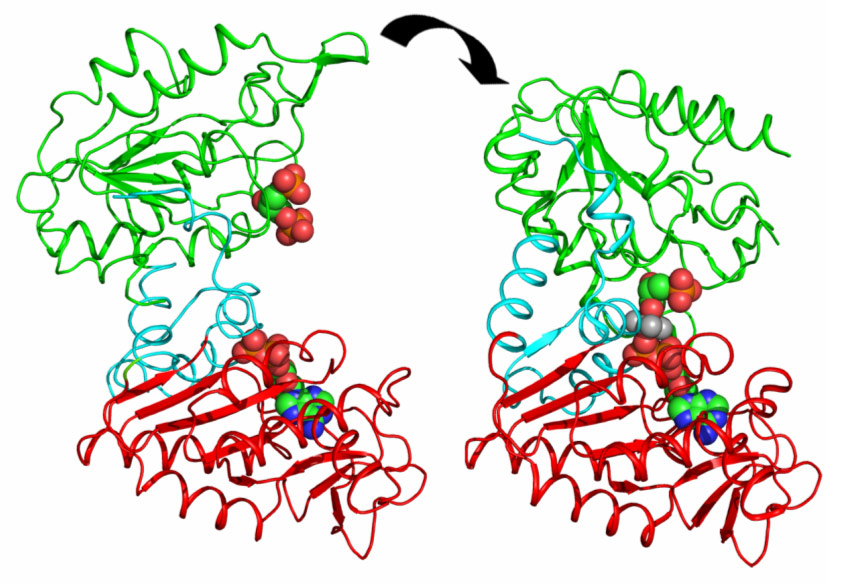
PGK is composed of two domains that bind two substrates separately (Figure 1). Early crystal structures already indicated that the open state of the enzyme was incapable of catalysis. The relative orientation of the two domains keeps the two substrates too distant from each other to allow phospho-transfer. Therefore, it was suggested that a significant closure of the enzyme via a “hinge-bending” mechanism is required for catalysis to occur. A major step forward in the understanding of catalysis by PGK was the determination of the fully closed crystal structures of the enzyme in complex with transition state analogues (TSAs) [1]. These defined, for the first time, the conformation of PGK required for its catalytic activity. However, the mechanism and extent of hinge-bending remained unknown.
 |
|
Figure 1. Domain movements in PGK catalysis. The fully-open resting state of the enzyme defined by DEN refinement against SAXS data (left) binds the substrates 13BPG in the N domain (green) and ADP in the C-domain (red). A rotation of 56° about a hinge region (blue) brings the substrates together to initialise catalysis and ATP production (right) (Image credit: M.W. Bowler). |
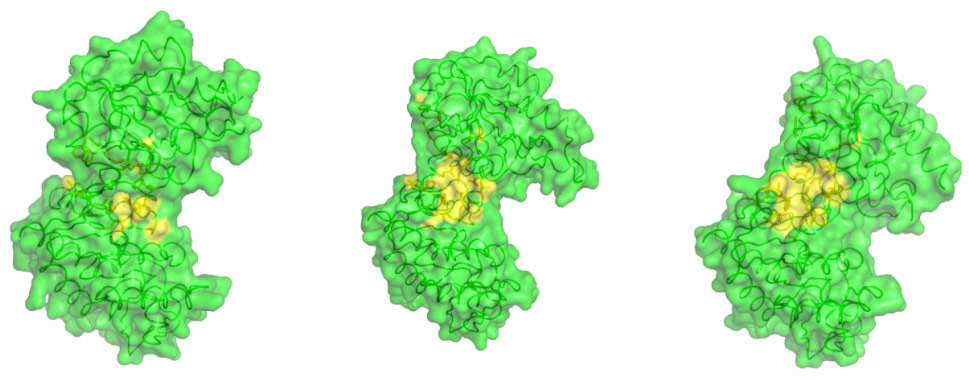
By combining high-resolution crystal structures with solution small-angle X-ray scattering (SAXS) experiments the extent and timing of the domain motions that lead to catalysis in PGK have now been elucidated. First, SAXS measurements were used to examine the resting, open state of the enzyme when no substrates are bound. By comparing the SAXS envelope determined for the protein in solution to crystal structures it was found that the enzyme adopts a more open conformation in solution than previously thought. Various states of the enzyme in catalytic turnover were then also investigated in solution. By comparing the crystal structures of the enzyme in different conformations to the solution scattering data obtained during catalytic turnover, the relative contribution of each state to the SAXS intensity distribution can be obtained. Surprisingly, it was found that, even in full catalytic turnover, the enzyme spends only 7% of its time in the fully-closed, catalytically-active conformation and remains in the fully-open conformation most of the time. To define the origin of the stability of the open conformation, a detailed model of the molecular interactions of this state was required. However, as SAXS data provide details to around only 1.5 nm resolution, these details are missing. By refining the atomic positions of a crystal structure against the SAXS data and applying deformable elastic network (DEN) protocols [2], a high resolution model of the fully open conformation observed in solution was obtained. Analysis of this model revealed that a large, solvent-exposed hydrophobic patch present in the closed conformation is masked from solvent in the open conformation (Figure 2). The occlusion of this hydrophobic region from the solvent could account for the preference of PGK to adopt an open conformation, as this will be thermodynamically more stable and implies that, while the enzyme constantly moves between the open and closed states, it has an energetic preference for the open conformation. The hydrophobic patch thus acts as a ‘spring’, constantly applying pressure to the closed conformation and leading to efficient release of products after catalysis. A model of cycling and catalysis was therefore proposed where the unliganded PGK remains open (ready to bind substrates, or release products) most of the time. The binding of substrates induces structural changes that allow the stabilisation of the closed state for sufficient time to enable catalysis. The interpolation of crystal structures and SAXS data has thus allowed a complete picture of the reaction cycle to be defined (see animations below).
 |
|
Figure 2. A hydrophobic spring regulates domain movement and catalysis in PGK. Surface representation of the transition from fully open to fully closed (left to right). Residues involved in hydrophobic interactions are coloured yellow. As the enzyme moves to the fully closed conformation a hydrophobic patch is exposed (Image credit: M. W. Bowler). |
A study of this kind required a completely integrated environment for Structural Biology. The crystallography and bio-SAXS measurements were carried out at the highly-automated ESRF Structural Biology beamlines ID14-2 and ID14-3. These two facilities enabled the investigation of large numbers of solutions using the EMBL/ESRF liquid handling robot and provided complementary techniques such as controlled crystal dehydration that were essential in the determination of crystal structures [3].
Animations
- Video 1 (above): The active site during the reaction cycle of human PGK
- Video 2: A hydrophobic spring regulates human PGK
- Video 3: The reaction cycle of human PGK
Principal publication and authors
L. Zerrad (a), A. Merli (b), G.F. Schröder (c), A. Varga (d), E. Graczer (d), P. Pernot (a), A. Round (e), M. Vas (d), and M.W. Bowler (a), A spring loaded release mechanism regulates domain movement and catalysis in phosphoglycerate kinase, J. Biol. Chem. 286, 14040-14048, (2011).
(a) ESRF
(b) University of Parma, Parma (Italy)
(c) Forschungszentrum Jülich, Jülich (Germany)
(d) Hungarian Academy of Sciences (Hungary)
(e) EMBL, Grenoble (France)
References
[1] M.J. Cliff et al., J. Am. Chem. Soc. 132, 6507-6516 (2010).
[2] G.F. Schröder et al., Nature 464, 1218-1222 (2010).
[3] S. Russi et al., J. Struct. Biol. (2011), doi:10.1016/j.jsb.2011.03.002.
Top image: Tracking the protein motions that harvest energy (Image credit: M. W. Bowler).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.