- Home
- News
- Spotlight on Science
- Silicon carbonate...
Silicon carbonate phase formed from carbon dioxide and silica under pressure
02-08-2011
The recent discovery of non-molecular silica-like carbon dioxide motivated a team of scientists from Italy and France to attempt to react carbon dioxide with silicon dioxide under high pressure. They created a new silicon carbonate phase by reacting a micro-porous SiO2 zeolite and molecular CO2 in a diamond anvil cell. Evidence of this new phase was provided by optical spectroscopy and synchrotron X-ray diffraction. A new oxide chemistry at high pressures and the potential for synthesis of a new class of materials are revealed, along with potential implications for CO2 segregation in planetary interiors and for CO2 storage.
Share
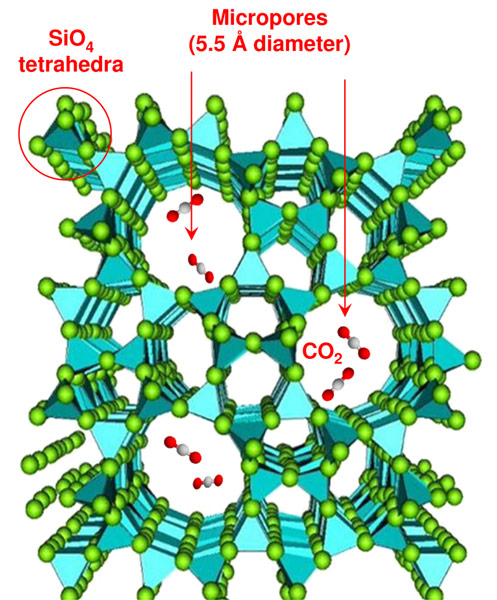
CO2 and SiO2 are two archetypal group IV oxides of paramount importance for chemistry and planetary sciences. The chemical relationship between these substances, in particular their reactivity, is of great interest. Although the two systems are both group IV oxides, they are remarkably different under ambient conditions as CO2 is molecular and is held together by C=O double bonds, while SiO2 forms network structures involving single Si-O bonds. These bonding patterns radically change under pressure. In fact, CO2 has been found to form non molecular solid phases above 30 GPa that bear similarities to the SiO2 system [1, 2]. A possible approach to favour the chemical reaction between CO2 and SiO2 is to select a microporous silica polymorph such as silicalite. At ambient conditions, silicalite is characterised by a framework of four-, five, six- and ten-membered rings of SiO4 tetrahedra with 5.5 Å pores [3], that can be completely filled by CO2 under pressure [4] (Figure 1). The choice of silicalite is driven by the large effective surface exposed to the CO2 in the pores, which is likely to be a crucial factor for enhancing the chemical reaction.
 |
|
Figure 1. SiO2, silicalite structure. Carbon dioxide fills the micro-pores of the zeolite under pressure, and reacts with the silica framework upon heating. |
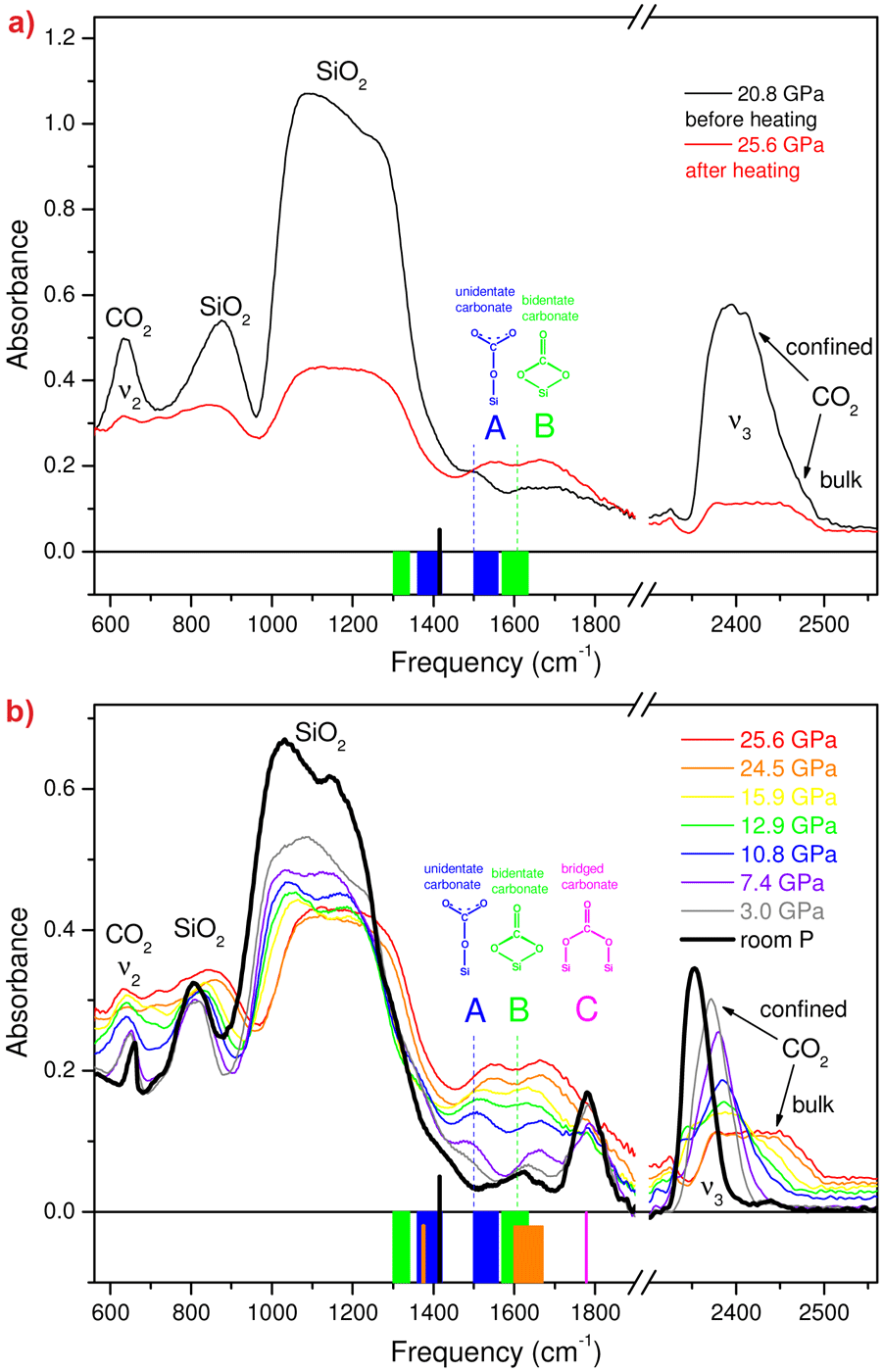
We examined chemical reactions between silicalite SiO2 and CO2 by compressing mixed samples at 18-26 GPa and then heating them up to 600-740 K. In the IR spectrum of the temperature quenched material (Figure 2a), the peaks of silicalite are reduced to 50% of the original intensity, and the peaks of micro-confined CO2 have almost completely vanished. In parallel, two new strong peaks A and B appear, that belong neither to molecular CO2 nor to silicalite. Therefore, the most fundamental aspect of a binary chemical reaction is demonstrated: two reactants, silicalite and CO2, react with each other, and a product is formed, identified by peaks A and B. Upon lowering the pressure the new peaks gradually disappear below 15 GPa and a new, non molecular peak, C, emerges at about 1780 cm-1. In parallel, we observe the formation of micro-confined CO2 and again an intensification of the silicalite peaks (Figure 2b). Finally, at room pressure, peak C disappears after a few days along with the peak of residual confined CO2, thereby showing the overall reversibility of the transformation. Peaks A, B and C were straightforwardly assigned to C-O stretching modes of different silicon carbonates (see the original publication), including unidentate and bidentate carbonates, both involving a single framework silicon atom, and bridged carbonates that involve two silicon atoms.
 |
|
Figure 2. IR spectra of mixed silicalite and CO2 showing the formation of silicon carbonate. The new compound is identified by peaks A, B, and C, assigned to unidentate, bidentate and bridged silicon carbonate species, respectively. a) IR spectra before and after heating to 740 K. After heating, the microconfined CO2 has almost vanished and the absorption of silicalite peaks decreases by about 50%. b) IR spectra measured upon decreasing pressure, after annealing. The behaviour of the ν3 CO2 peak shows that confined CO2 forms again (sharp component), starting at about 16 GPa while the signature of bulk CO2 (broad component) vanishes, indicating that this material progressively extrudes from the sample chamber. Vertical black bars: frequency of the IR stretching mode of the free CO3 ion. Blue, green and orange bars: spectral ranges, at ambient pressure, of the split high and low frequency components of the IR stretching mode of the free CO3 ion, as found in literature in unidentate (blue), bidentate (green) and bridging, T-CO3-T (orange) carbonates, obtained from CO2 adsorption on metal oxides and dissolution in silicate melts. Pink bar (only in b): spectral range, at ambient pressure, for the highest frequency IR stretching mode of bridging carbonates also obtained from adsorption of CO2 on some metal oxides. Vertical, dashed lines: frequencies of peaks A and B extrapolated to room temperature. |
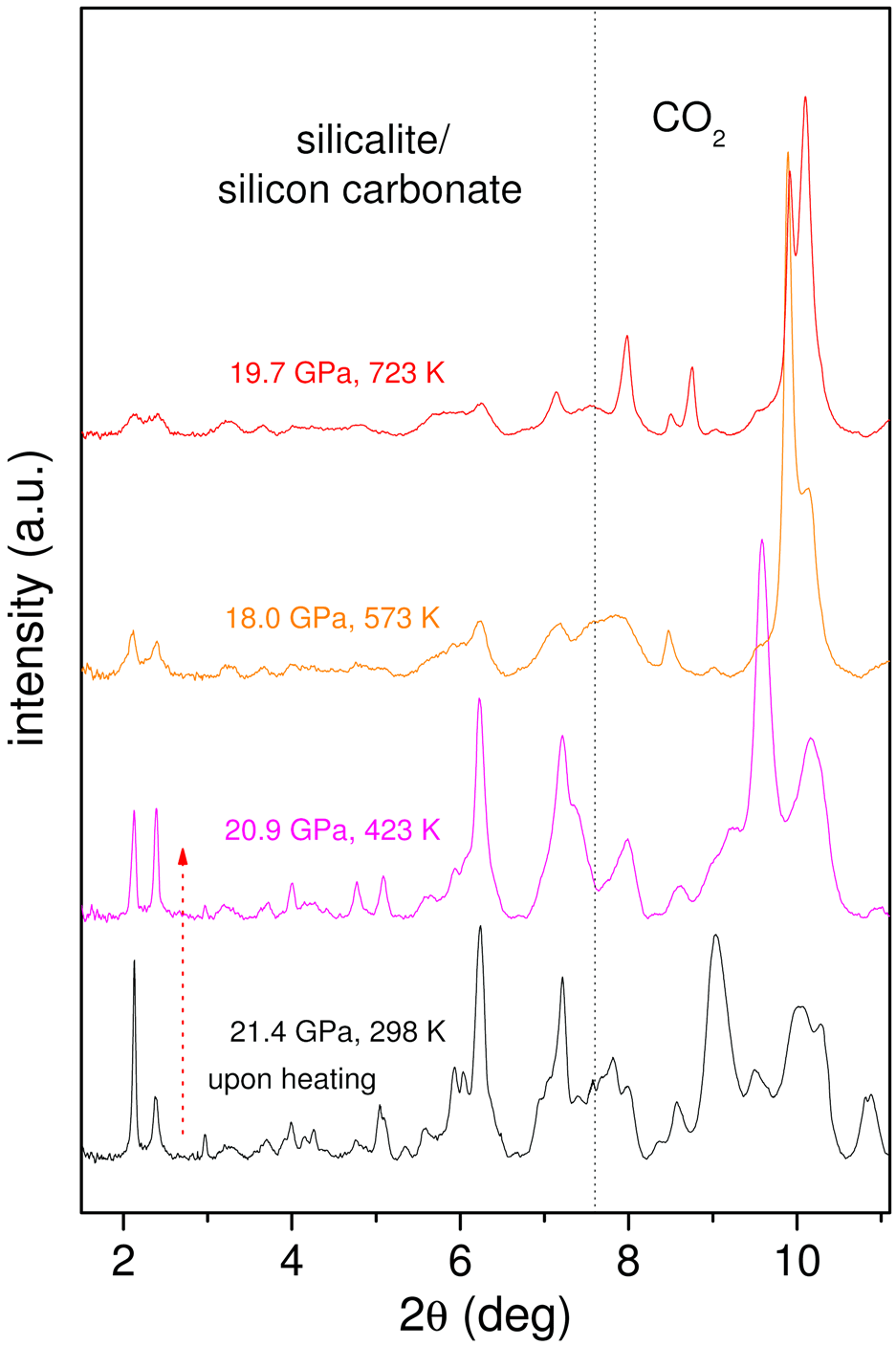
As an additional test of the chemical reaction and of the nature of the compound, we measured the X-ray diffraction (XRD) patterns (Figure 3) at beamline ID27 on a sample compressed to 21.4 GPa and then heated to 723 K. The Bragg peaks of orthorhombic silicalite progressively broaden upon increasing temperature. This is a clear indication of the chemical reaction, as diffraction peaks should instead sharpen with increasing temperature T because of the usual temperature enhancement of the crystal quality and relaxation of any deviatoric stress present. It was therefore shown that the chemical reaction takes place progressively, resulting in a product that is a highly-strained crystal still exhibiting the structure of the original silicalite. This is not unexpected since the carbonates form at the micropore surface, which in turn does not alter the pore arrangement within the unit cell, but affects the long-range periodicity of the structure. The carbonate groups form in a random manner without any long-range order. They can also be expected to induce local geometrical distortions to the framework.
 |
|
Figure 3. High pressure XRD patterns of a silicalite-CO2 mixture showing the formation of a highly-strained, disordered crystal upon heating. At angles higher than the vertical dotted line, the diffraction pattern is dominated by the peaks of bulk molecular CO2, that undergoes phase transitions between phases I, II, III and IV. |
The data presented here show that SiO2 and CO2 undergo high P-T chemical reactions of the type: xSiO2+yCO2 « SixCyO(2x+2y), that result in the formation of one or more silicon carbonate compounds. Although the reaction occurs at the surface of the silicalite micropores, the final product has the nature of a real bulk compound due to the particular structure of zeolitic silicalite. All of the tetrahedra in silicalite are on the surface of the micropores, thus a surface reaction can involve, in principle, all the SiO2, as would be the case in a bulk reaction. However, the silicalite framework is retained, although highly strained, and the product is thus a non-stoichiometric silicon carbonate.
Principal publication and authors
Silicon carbonate phase formed from carbon dioxide and silica under pressure, M. Santoro (a,b), F.A. Gorelli (a,b), J. Haines (c), O. Cambon (c), C. Levelut (d), G. Garbarino (e), Proc. Natl. Acad. Sci. U.S.A. 108, 7689-7692 (2011).
(a) European Laboratory For Non Linear Spectroscopy (LENS), Firenze (Italy)
(b) IPCF-CNR, Roma (Italy)
(c) Institut Charles Gerhardt Montpellier, Equipe Chimie et Cristallochimie des Matériaux, UMR 5253, CNRS, Université Montpellier 2 (France)
(d) Laboratoire Charles Coulomb, UMR 5221, CNRS, Université Montpellier 2 (France)
(e) ESRF
References
[1] M. Santoro, and F.A. Gorelli, Chem. Soc. Rev. 35, 918-931 (2006).
[2] M. Santoro, et al., Nature 441, 857-860 (2006).
[3] J. Haines, et al., J. Am. Chem. Soc. 131, 12333-12338 (2009).
[4] J. Haines, et al., J. Am. Chem. Soc. 132, 8860-8861 (2010).
Top image: Are compounds between carbon dioxide and silicon dioxide possible?
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.