MASSIF-1 FAQ
Answers to some Frequently Asked Questions about MASSIF-1
1. How can I cite MASSIF-1?
If you found MASSIF-1 useful for screening, data collection and/or structure solution please cite the following papers:
Svensson, O., Gilski, M., Nurizzo, D. & Bowler, M. W. (2018). Multi-position data collection and dynamic beam sizing: recent improvements to the automatic data-collection algorithms on MASSIF-1, Acta Cryst. D74, 433-440, http://dx.doi.org/10.1107/S2059798318003728
- Describes the improvements made to the automatic data collection algorithms including dynamic adaptation of beam diameter to crystal volume and multi-position and helical data collection (MXPressP)
Svensson, O., Monaco, S., Popov, A. N., Nurizzo, D. & Bowler, M. W. (2015). The fully automatic characterization and data collection from crystals of biological macromolecules, Acta Cryst. D71, 1757-1767, http://dx.doi.org/10.1107/S1399004715011918
- Describes the original developments in sample location, characterisation and data collection algorithms (automesh, X-ray centring, MXPressE and SAD data collection protocols etc.), should be cited for all experiments conducted on MASSIF-1
Bowler M.W., Nurizzo, D., Barrett, R., Beteva, A., Bodin, M., Caserotto, H., Delageniere, S., Dobias, F., Flot, D., Giraud, T., Guichard, N., Guijarro, M., Lentini, M., Leonard, G., McSweeney, S., Oskarsson, M., Schmidt, W., Snigirev, A., von Stetten, D., Surr, J., Svensson, O., Theveneau, P. and Mueller-Dieckmann, C. (2015) MASSIF-1: A beamline dedicated to the fully automatic characterisation and data collection from crystals of biological macromolecules J. Sync. Rad. 22 1540-1547 http://dx.doi.org/10.1107/S1600577515016604.
- Describes the technical layout of the beamline, should be cited for all experiments conducted on MASSIF-1
Bowler, M.W., Svensson, O. & Nurizzo, D. (2016): Fully automatic macromolecular crystallography: the impact of MASSIF-1 on the optimum acquisition and quality of data, Cryst. Rev., 22, 233–249 http://dx.doi.org/10.1080/0889311X.2016.1155050.
- Describes the first full year of operation of MASSIF-1, analysing the results from the large scale automation of MX data collection showing the advantages of fully autonomous data collection
Nurizzo, D., Bowler M.W., Caserotto, H., Dobias, F., Giraud, T., Surr, J., Guichard, N., Papp, G., Guijarro, M., Mueller-Dieckmann, C., Flot, D., McSweeney, S. Cipriani, F, Theveneau, P. and Leonard, G. (2016) RoboDiff: combining a sample changer and goniometer for highly automated macromolecular crystallography experiments Acta Cryst D 72, 966-975, http://dx.doi.org/10.1107/S205979831601158X.
- Describes the core robotic automation of the beamline
2. Do you pay for dewar shipping?
Yes! As of June 2017 the ESRF will pay all transport costs for 1 dewar per shift (3 dewars for 24 hours) for academic time allocated on MASSIF-1. Please click here for details.
3. Do you take unipucks?
Yes! From September 2020 MASSIF-1 is only compatible with unipucks.
4. How can I book time on MASSIF-1?
Time on MASSIF-1 is not scheduled in the same way as the other MX beamlines. Users that are part of a BAG can book time, in consultation with their BAG responsible, on the MASSIF-1 Google calander. When the samples arrive they enter a queuing system and results are guaranteed within 3 working beam days of the allotted slot. The number of samples processed on the beamline is then converted into shifts and counted against allocated beamtime for the BAG. Applications can also be made via a rolling process and once shifts have been allocated slots are booked on-line in the same manner as for BAGs.
5. It is the day I booked but data collection has not started
We have a flexible booking and data collection system - this means that data collection may not occur on the day you booked in order to stream line the process or deal with technical problems. We guarentee data collection within 3 working beam days of your selected date and an email is automatically triggered when data collection starts
6. Can I contact someone to help?
Yes! Please contact Matthew Bowler if you would like to discuss how best to use the beamline, need advice for your samples or if you have suggestions to improve or modify the way we treat samples.
7. My sample is not centred properly!
If there is very weak diffraction (a DOZOR score below 1) it often looks like the pin is centered or the loop is badly centred. If you look at the automesh image (found by clicking the MXPressE link in the ISPyB summary for a sample then clicking on the "Results" tab, see example below) you can verify that the mesh covered the loop, but as there was no diffraction it centred on noise. We prefer to leave the centring routine to continue as some crystals do have a very weak signal and data can be collected anyway - if you inspect the raw images from the mesh you will see that there was very very weak or no diffraction from these crystals. The comments are filled in ISPyB with "Very weak diffraction" for DOZOR scores below 1

Result of the automesh - the red box defines the area of the mesh scan so you can verify that the sample mount was scanned correctly.
8. What is the diffraction plan and how can I use it?
The diffraction plan holds all the information needed on a sample and can be described in ISPyB. Essential entries in the diffraction plan are the sample acronym and a unique sample name as well as the puck barcode.
Additional information can be added to tailor the experiment performed for each sample:
| Diffraction-plan entry | Definition | Default value |
|---|---|---|
| Protein acronym | Defines the protein that is registed with the ESRF safety group (SMIS) | Required field |
| Sample name | User-defined unique identifier | Required field |
| Pin barcode | Barcode identifier | None |
| Experiment type | Define MXPressE/O/SAD/Score/M/P/P_SAD/F | MXPressE |
| Crystal form | If present (from an associated PDB) used for strategy calculation and autoprocessing (except some Grenades jobs) | None |
| Aimed resolution | Resolution that the detector will be set to for mesh scans, characterization images and default data collection | 2.0 Å |
| Required resolution | Threshold resolution; samples below the cutoff will not be collected | None |
| Radiation-sensitivity | BEST input in the case of highly radiation-sensitive crystals (0.5–2.0 - low to high sensitivity) | 1 |
| Required completeness | % of Completeness required (value between1 and100) | 99% |
| Required multiplicity | — | 4 |
| No. of positions | For multiple crystals on your support or for helical data collection (MXPressP) | 1 (5 for MXPressP and P_SAD) |
| Beam diameter | Select appropriate beam size for crystals (if blank will be dynamically adapted to crystal size) - value in µm | 50 µm |
| Forced Space Group | Select space group for strategy calculation and autoprocessing if cell not known or no PDB | None |
| Total rotation angle | Select required rotation range for data collection |
MXPressE - minimum required MXPressE_SAD - 360° MXPressO - 180° MXPressF - 180° MXPressP - 180°/positions MXPressP_SAD - 360°/positions |
| Observed resolution & Comments are two fields that are not used by our Worklows, but are only a help to the users | ||
The diffraction plan interface in ISPyB:
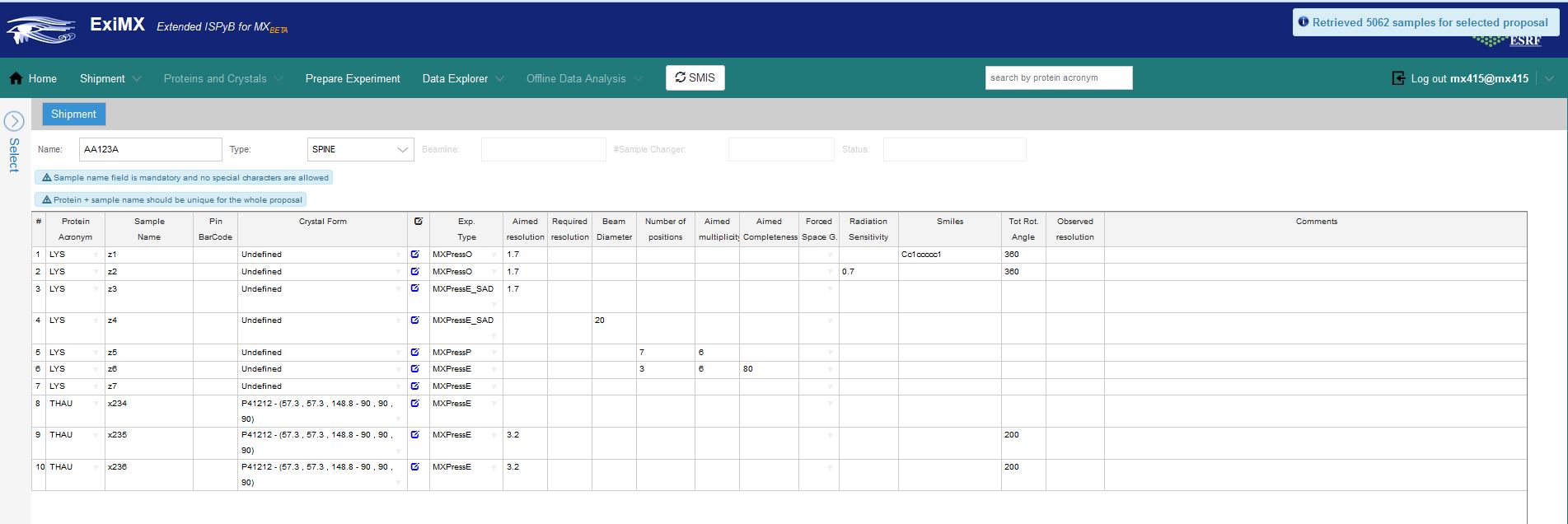
The create puck interface in ISPyB is where information about each sample is uploaded. The example above shows a mixture of samples where information has been added.
The only compulsory fields are sample acronym and name and the puck barcode (in the 'Puck:' field above).
All other fields are optional and have sensible default values if no information is provided (see table above). The experiment type field is important as if an experiment other than MXPressE is required (such as MXPressE_SAD, SCORE or MXPressO, click here for more details) it must be entred here. The beamsize is an option to select the diameter of the beam in microns - this should be appropriate to your crystals. All other fields contribute to the strategy for data collection where the space group can be forced, resolution, multiplicity etc can be chosen.
9. Someone has booked in samples already for a day in the Google calander, can I book some as well?
Yes! As long as the total number of crystals does not exceed 150 for 24 hours multiple groups can add crystals.
10. Where are my images?
The best place to look at results is in ISPyB - here you can also download processed data. Otherwise everything is in the directories with the following structure:
/data/visitor/mx1234/id30a1/20150631/RAW_DATA/Acronym/Sample_Name
and
/data/visitor/mx1234/id30a1/20150631/PROCESSED_DATA/Acronym/Sample_Name
All mesh, line and characterisation images are in a sub-directory for each sample called /MXPressE_01
All data sets will be in the directory /data/visitor/mx1234/id30a1/20150631/RAW_DATA/Acronym/Sample_Name
11. I asked for a MXPressE SAD strategy and my processed data have very low completeness at high resolution
The SAD strategy chooses the resolution to collect where the Rmerge between Bijvoet pairs is 5%. This is the resolution where there should be significant anomalous signal but the autoprocessing pipelines will process to where the signal to noise is reasonable - this is much higher resolution than that selected for SAD leading to low completeness.
12. Why are some of my samples missing after the run?
There are 2 scenarios when we put samples in the 'poubelle':
1. After data collection if the pin is not pick up properly or there is a collision in the dewar between the gripper and the puck
2. Sometimes the robot takes a sample out of the dewar with wrong orientation and the robot cannot realign it automatically.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.